Phenylketon niệu là một chứng rối loạn di truyền hiếm gặp gây ra bởi sự thay đổi gen phenylalanine hydroxylase (PAH) khiến một loại axit amin gọi là phenylalanine tích tụ trong cơ thể.
Nội dung:
- 1 Bệnh Phenylketon niệu là gì?
- 2 Đặc điểm dịch tế học của bệnh Phenylketon niệu
- 3 Đặc điểm sinh lý bệnh của Phenylketon niệu
- 4 Phân loại bệnh Phenylketon niệu
- 5 Sự thiếu hụt enzyme Phenylalanine hydroxylase
- 6 Chẩn đoán bệnh Phenylketon niệu
- 7 Điều trị bệnh Phenylketon niệu
- 8 Bệnh Phenylketon niệu ở mẹ bầu (Phenylalanine Embryopathy)
- 9 Tài liệu tham khảo
Bệnh Phenylketon niệu là gì?
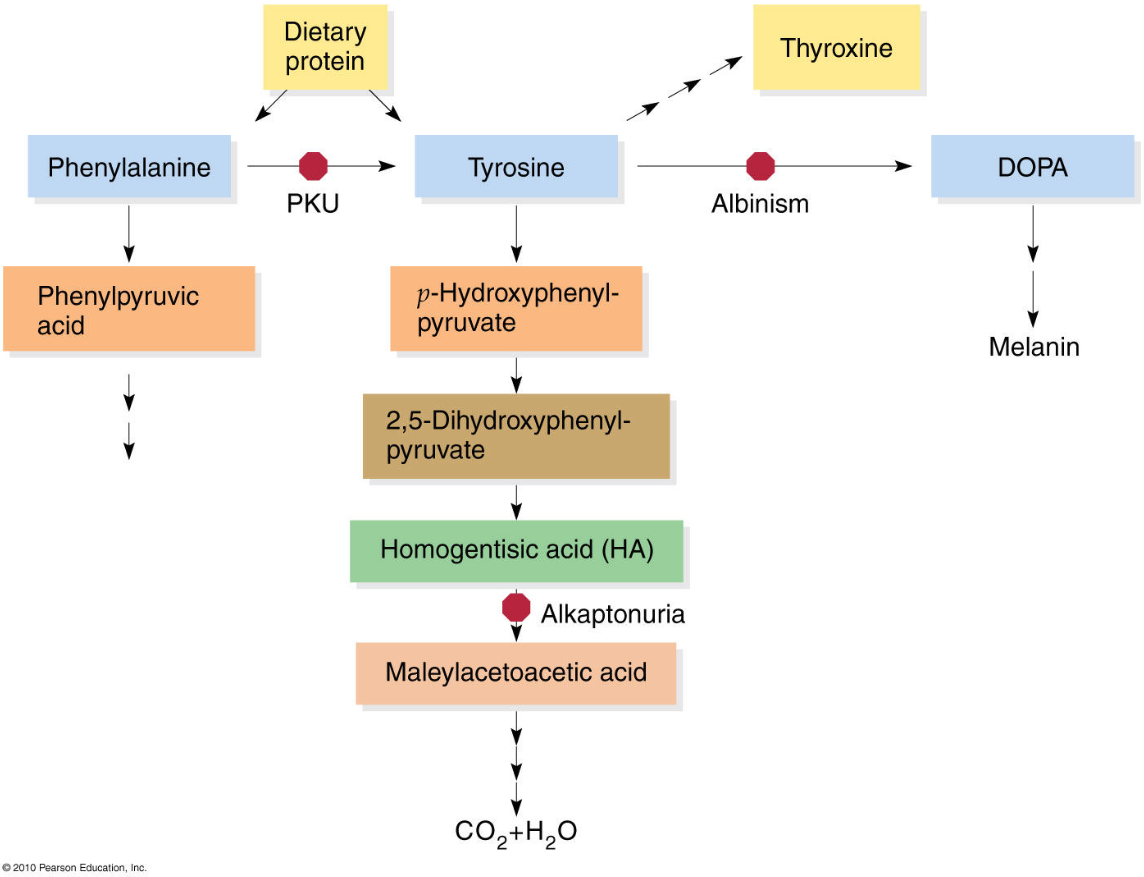
Phenylketon niệu (Phenylketonuria, PKU, MIM #261600) là một chứng rối loạn di truyền hiếm gặp gây ra bởi sự thay đổi gen mã hóa phenylalanine hydroxylase (PAH) khiến một loại axit amin gọi là phenylalanine tích tụ trong cơ thể. Gen PAH giúp tạo ra enzym cần thiết để phân hủy phenylalanine.
Kết quả của sự thiếu hụt phenylalanine hydroxylase (PAH) và nếu không được điều trị sẽ dẫn đến tình trạng thiểu năng trí tuệ không hồi phục cùng với các triệu chứng lâm sàng khác.
Đặc điểm dịch tế học của bệnh Phenylketon niệu
Tỷ lệ mắc PKU là khoảng 1/10.000 ở dân số châu Âu, mặc dù bệnh này ít phổ biến hơn ở dân số người Mỹ gốc Phi, với tỷ lệ mắc khoảng 1/50.000.
PKU rất hiếm ở Phần Lan; và cũng hiếm gặp ở Nhật Bản, mặc dù tỷ lệ mắc bệnh có thể khác nhau rõ rệt giữa các vùng khác nhau. Hầu hết các trường hợp PKU là do thiếu hụt phenylalanine hydroxylase (PAH).
Đặc điểm sinh lý bệnh của Phenylketon niệu
Enzyme gan phenylalanine hydroxylase (PAH) xúc tác quá trình chuyển đổi axit amin thiết yếu phenylalanine thành tyrosine.
Tetrahydrobiopterin (BH4) là một đồng yếu tố cần thiết cho hoạt động của PAH ngoài oxy phân tử và sắt. Con đường này chiếm phần lớn quá trình dị hóa và chịu trách nhiệm loại bỏ khoảng 75% phenylalanine trong chế độ ăn uống, phần còn lại được sử dụng để tổng hợp protein.
PKU là do thiếu PAH. Điều này dẫn đến nồng độ phenylalanine trong máu và nước tiểu và các chất chuyển hóa của nó, phenylacetate và phenyllactate tăng cao.
Nồng độ tyrosine thường ở mức bình thường, mặc dù đôi khi cũng quan sát thấy nồng độ thấp. Khiếm khuyết trong chuyển hóa BH4 chiếm khoảng 2% bệnh nhân có nồng độ phenylalanine tăng cao.
Phân loại bệnh Phenylketon niệu
Dựa vào mức độ sản sinh enzyme mà bệnh PKU có thể được phân loại ở các mức độ khác nhau:
* PKU cổ điển: Dạng rối loạn nghiêm trọng nhất được gọi là PKU cổ điển. Enzyme cần thiết để phân hủy phenylalanine bị thiếu hoặc giảm nghiêm trọng. Nồng độ phenylalanine trong huyết thanh ở trẻ sơ sinh mới được chẩn đoán, chưa được điều trị vượt quá 20 mg/dL (1200 micromol/L). Điều này dẫn đến lượng phenylalanine cao có thể gây tổn thương não nghiêm trọng.
* Các dạng PKU ít nghiêm trọng hơn: Ở dạng nhẹ hoặc trung bình, enzyme này vẫn có một số chức năng, do đó nồng độ phenylalanine không cao, dẫn đến nguy cơ tổn thương não đáng kể ít hơn. Các ngưỡng nồng độ khác của enzyme gây ra dạng bệnh PKU vừa phải (nồng độ phenylalanine từ 900 đến 1200 micromol/L), PKU nhẹ (nồng độ phenylalanine từ 600 đến 900 micromol/L), chứng tăng phenylalanin huyết nhẹ (HPA; nồng độ phenylalanine từ 360 đến 600 micromol/L) và HPA nhẹ lành tính thường không cần xử lý (nồng độ phenylalanine từ 120 đến 360 micromol/L).
Bất kể ở dạng nào, hầu hết trẻ sơ sinh, trẻ em và người lớn mắc chứng rối loạn này vẫn cần một chế độ ăn kiêng PKU đặc biệt để ngăn ngừa thiểu năng trí tuệ và các biến chứng khác.
Cơ chế của triệu chứng thiểu năng trí tuệ
Cơ chế mà nồng độ phenylalanine tăng cao gây ra thiểu năng trí tuệ phần lớn chưa được biết rõ. Quá nhiều phenylalanine được cho là cản trở sự phát triển của não, quá trình myel hóa và tổng hợp chất dẫn truyền thần kinh.
Các cơ chế tiềm năng đã được đề xuất dựa trên các nghiên cứu sau:
* Sự phát triển bất thường của tế bào thần kinh, thần kinh đệm và ma trận ngoại bào của não ở những con chuột sau khi sinh tiếp xúc với nồng độ phenylalanine cao được cho là do những thay đổi trong các dấu hiệu phân tử. Chúng bao gồm tăng hoạt động liên kết với hyaluronate trong quá trình hình thành ma trận ngoại bào, thay đổi thành phần của phân tử kết dính tế bào thần kinh và tăng protein axit dạng sợi thần kinh đệm trong quá trình phát triển tiểu não.
* Nồng độ phenylalanine cao có thể làm gián đoạn sự phát triển của não thông qua stress oxy hóa: Trong não của những con chuột mắc chứng hyperphenylalaninemia, tổng tiềm năng chống oxy hóa bẫy gốc tự do đã giảm và phát quang hóa học tăng lên. Phenylalanine ức chế cả catalase và glutathione peroxidase in vivo. Tuy nhiên, catalase chỉ bị ức chế trong ống nghiệm và superoxide dismutase không bị ảnh hưởng trong cả hai tình trạng.
* Các chất chuyển hóa phenylalanine, phenylacetate và phenylpyruvate, có thể ức chế tổng hợp alpha-tocopherolquinone, một đồng yếu tố thiết yếu để tổng hợp arachidonic não và axit docosahexaenoic, axit béo không bão hòa đa cần thiết cho sự phát triển bình thường của não.
* Trong các tế bào hồi hải mã của chuột được nuôi cấy, phenylalanine đặc biệt ức chế các thụ thể N-methyl-D-aspartate (NMDA), được cho là có liên quan đến việc điều chỉnh trí nhớ và học tập.
* Việc vận chuyển các axit amin trung tính lớn (LNAAs) vào não bị ức chế do nồng độ phenylalanine tăng lên: LNAA giảm được cho là ức chế quá trình tổng hợp protein và chất dẫn truyền thần kinh, dẫn đến thiếu hụt nồng độ dopamine và serotonin.
Sự thiếu hụt enzyme Phenylalanine hydroxylase
Mô hình di truyền của bệnh Phenylketon niệu là di truyền lặn trên nhiễm sắc thể thường.
Gần như tất cả các trường hợp đều do đột biến gen mã hóa phenylalanine hydroxylase (PAH), đã được xác định vị trí trên nhiễm sắc thể số 12 người ở tọa độ 12q24.1
Hơn 1000 loại đột biến, bao gồm xóa, chèn, khiếm khuyết nối và đột biến tên lửa và vô nghĩa, có liên quan đến tình trạng thiếu PAH.
Không có đột biến đơn lẻ nào chiếm ưu thế ở người da trắng, mặc dù một số đột biến nhất định phổ biến hơn ở các nhóm dân tộc cụ thể. Hầu hết các bệnh nhân bị ảnh hưởng là dị hợp tử đối với hai đột biến khác nhau.
Đột biến ảnh hưởng đến cấu trúc của PAH.
PAH là một tetramer, với mỗi monome bao gồm miền xúc tác và miền tetramer hóa.
Hầu hết các đột biến dẫn đến PKU đều nằm trong vùng xúc tác. Tuy nhiên, một số xảy ra ở khu vực giao thoa của hai vùng, nơi chúng có thể ảnh hưởng đến tính ổn định của enzyme.
Các đặc điểm lâm sàng
Do các kỹ thuật sàng lọc sơ sinh đã được triển khai rộng rãi nên các biểu hiện lâm sàng rõ ràng của PKU rất hiếm. Trẻ sơ sinh không có triệu chứng trước khi bắt đầu cho ăn thức ăn có chứa phenylalanine (ví dụ: sữa mẹ hoặc sữa bột tiêu chuẩn dành cho trẻ sơ sinh).
Nếu không được phát hiện trong giai đoạn sơ sinh, sự khởi phát của PKU diễn ra âm thầm và có thể không gây ra các triệu chứng cho đến khi trẻ ở độ tuổi sơ sinh.
Ở những bệnh nhân không được điều trị, dấu hiệu đặc trưng của bệnh là thiểu năng trí tuệ không hồi phục, co giật, bất thường về hành vi, tật đầu nhỏ và bệnh về da (chàm phát ban, sắc tố nhẹ) do tăng phenylalanin huyết (HPA).
Các phát hiện khác bao gồm những bất thường về dáng đi, tư thế ngồi và thế đứng. Cơ thể và nước tiểu có thể có mùi đặc trưng do nồng độ axit phenylacetic tăng lên.
Nếu không có chế độ ăn kiêng hạn chế, tình trạng suy giảm nhận thức trở nên tồi tệ hơn trong quá trình myel hóa ở thời thơ ấu với việc gia tăng tiếp xúc với phenylalanine trong chế độ ăn uống nhưng sẽ ổn định khi quá trình trưởng thành của não bộ hoàn tất.
Một nghiên cứu gồm 51 bệnh nhân không được điều trị được đánh giá ở độ tuổi từ 28,8 đến 71,8, mức độ thiểu năng trí tuệ được chia gần như đồng đều giữa mức độ nghiêm trọng (chỉ số thông minh [IQ] <35) và mức độ suy giảm nhẹ đến trung bình (IQ 36 đến 67). Hai người có IQ >68. Động kinh xảy ra thường xuyên, với 12 (23%) bệnh nhân trưởng thành bị ảnh hưởng. Một số bệnh nhân bị mất chức năng vận động theo thời gian, mặc dù 41 (80%) không bị suy giảm thêm.
Bệnh nhân được điều trị bằng can thiệp chế độ ăn uống liên tục sau khi chẩn đoán sàng lọc sơ sinh vẫn có thể biểu hiện một số di chứng thần kinh, mặc dù ít nghiêm trọng hơn nhiều so với bệnh nhân không được điều trị. Một số khả năng nhận thức bị ảnh hưởng đặc biệt là chức năng điều hành (thùy trán). Suy giảm nhẹ ở khu vực này được thấy ngay cả ở những bệnh nhân kiểm soát tốt lượng phenylalanine trong chế độ ăn uống.
Một phân tích tổng hợp của 11 nghiên cứu bao gồm 360 bệnh nhân đã chứng minh rằng việc giảm mật độ khoáng của xương không có ý nghĩa lâm sàng và không có mối tương quan giữa mật độ khoáng của xương và nồng độ phenylalanine, lượng chất dinh dưỡng, vitamin D hoặc hormone tuyến cận giáp (Parathyroid Hormone, PTH).
Hình ảnh não của bệnh nhân Phenylketon niệu
Hình ảnh thần kinh không được thực hiện thường xuyên ở những bệnh nhân mắc PKU mà được dành riêng cho những người có biểu hiện thần kinh bất thường.
Tổn thương chất trắng rõ ràng trên hình ảnh cộng hưởng từ (MRI) ở phần lớn bệnh nhân mắc PKU, bao gồm cả những người được phát hiện qua sàng lọc sơ sinh và được điều trị bằng chế độ ăn kiêng sớm.
Một phát hiện phổ biến là sự gia tăng đối xứng tín hiệu T2W ở chất trắng quanh não thất. Những thay đổi này được cho là đại diện cho sự luân chuyển myelin tăng lên do nồng độ phenylalanine tăng lên và có thể hồi phục.
Mức độ nghiêm trọng của những bất thường dường như có liên quan đến tình trạng chế độ ăn uống. Điều này đã được chứng minh bằng một loạt 34 bệnh nhân PKU từ 8 đến 33 tuổi, trong đó 25 người được phát hiện bằng sàng lọc sơ sinh. Ở nhóm được chẩn đoán sớm, mức độ nghiêm trọng của những thay đổi trên MRI có liên quan đáng kể với nồng độ phenylalanine huyết thanh tại thời điểm nghiên cứu.
Chẩn đoán bệnh Phenylketon niệu
Chẩn đoán PKU dựa trên việc phát hiện nồng độ phenylalanine trong huyết thanh tăng cao, sau đó là xét nghiệm phân tử. Mức tăng trong máu có thể rất cao (>20 mg/dL, 1200 micromol/L) ở những bệnh nhân bị thiếu hoàn toàn PAH.
Phương pháp hữu ích nhất để sàng lọc trẻ sơ sinh là khối phổ song song.
Kỹ thuật này cũng có thể đo các axit amin bổ sung bao gồm tyrosine và acylcarnitine ester. Nồng độ phenylalanine cao cùng với nồng độ tyrosine thấp đến thấp bình thường gợi ý chẩn đoán PKU. Ngoài ra, phép đo khối phổ song song có thể xác định nhiều lỗi chuyển hóa bẩm sinh khác trong một mẫu. Nồng độ phenylalanine tăng cao được xác định thông qua sàng lọc sơ sinh nên được xác nhận bằng phân tích axit amin huyết tương thứ hai.
Phân tích enzyme không được thực hiện để xác nhận chẩn đoán, vì hoạt tính PAH chỉ được biểu hiện ở gan.
Chẩn đoán bằng kỹ thuật sinh học phân tử có thể được sử dụng để xác nhận chẩn đoán bệnh PKU thông qua việc xác định hai đột biến gây bệnh trong gen PAH (đột biến dị hợp tử hoặc đồng hợp tử).
Xét nghiệm di truyền có thể được sử dụng để phát hiện người mang mầm bệnh hoặc chẩn đoán trước sinh ở những gia đình đã biết (các) đột biến. Trong một số trường hợp, có thể dự đoán hoạt động của enzyme và/hoặc khả năng đáp ứng với tetrahydrobiopterin (BH4) từ kiểu gen PAH, mặc dù mối liên quan có thể không nhất quán.
Điều trị bệnh Phenylketon niệu
Bắt đầu điều trị sớm bệnh PKU và tiếp tục điều trị trong suốt cuộc đời có thể giúp ngăn ngừa thiểu năng trí tuệ và các vấn đề sức khỏe nghiêm trọng.
Các phương pháp điều trị chính cho PKU bao gồm:
- Một chế độ ăn uống suốt đời với lượng thức ăn rất hạn chế với phenylalanine.
- Dùng công thức PKU — một chất bổ sung dinh dưỡng đặc biệt — suốt đời để đảm bảo rằng bạn nhận đủ chất đạm thiết yếu (không có phenylalanine) và các chất dinh dưỡng cần thiết cho sự tăng trưởng và sức khỏe tổng quát. Một lượng phenylalanine an toàn khác nhau đối với mỗi người bị PKU và có thể thay đổi theo thời gian. Nói chung, ý tưởng là chỉ tiêu thụ lượng phenylalanine cần thiết cho sự phát triển lành mạnh và các quá trình của cơ thể, nhưng không nhiều hơn.
- Thuốc đặc trị cho bệnh PKU.
Hạn chế ăn thực phẩm giàu phenylalanine
Phương pháp điều trị chính trong PKU là hạn chế phenylalanine trong chế độ ăn uống.
Vì lượng phenylalanine mà một người bị PKU có thể ăn một cách an toàn quá thấp nên điều quan trọng là phải tránh tất cả các loại thực phẩm giàu protein, bao gồm:
- Sữa
- Trứng
- Phô mai
- Quả hạch
- Các sản phẩm đậu nành, chẳng hạn như đậu nành, đậu phụ, tempeh
- Đậu và đậu Hà Lan
- Gia cầm, thịt bò, thịt lợn và bất kỳ loại thịt nào khác
- Cá
- Khoai tây, ngũ cốc và các loại rau khác có thể sẽ bị hạn chế.
Trẻ em và người lớn cũng cần tránh một số loại thực phẩm và đồ uống khác, bao gồm nhiều loại soda ăn kiêng và đồ uống khác có chứa aspartame (NutraSweet, Equal). Aspartame là một chất làm ngọt nhân tạo được làm bằng phenylalanine.
Một số loại thuốc có thể chứa aspartame và một số vitamin hoặc chất bổ sung khác có thể chứa axit amin hoặc sữa bột tách béo. Kiểm tra với dược sĩ tại quầy thuốc về thành phần của các sản phẩm không kê đơn và thuốc kê đơn.
Công thức PKU
Do chế độ ăn kiêng hạn chế, những người bị PKU cần nhận được các chất dinh dưỡng thiết yếu thông qua một chất bổ sung dinh dưỡng đặc biệt. Công thức không chứa phenylalanine cung cấp protein thiết yếu (axit amin) và các chất dinh dưỡng khác ở dạng an toàn cho người bị PKU.
Công thức PKU là các loại thực phẩm dinh dưỡng y học bao gồm các chất thay thế protein không chứa phenylalanine (hỗn hợp axit amin) cung cấp khoảng 75% nhu cầu protein (ngoại trừ phenylalanine) dành riêng cho các bệnh nhân PKU.
Thực phẩm dinh dưỡng y học có thể chứa glycomacropeptide (GMP) làm nguồn protein. GMP là một loại protein tự nhiên có trong váng sữa phô mai có chứa một lượng nhỏ phenylalanine và được bổ sung một số axit amin trung tính lớn (LNAAs). Các nghiên cứu đã chứng minh tính hiệu quả và ngon miệng của chế độ ăn kiêng GMP ở bệnh nhân PKU.
* Công thức cho trẻ sơ sinh và trẻ mới biết đi: Bởi vì sữa công thức dành cho trẻ sơ sinh thông thường và sữa mẹ có chứa phenylalanine, thay vào đó, trẻ sơ sinh bị PKU cần phải dùng sữa công thức không chứa phenylalanine. Chuyên gia dinh dưỡng có thể tính toán cẩn thận lượng sữa mẹ hoặc sữa công thức thông thường được thêm vào công thức không chứa phenylalanine. Chuyên gia dinh dưỡng cũng có thể hướng dẫn cha mẹ cách chọn thức ăn đặc mà không vượt quá lượng phenylalanine cho phép hàng ngày của trẻ.
Trẻ sơ sinh bị PKU đang bú sữa mẹ được khuyến khích dưới sự giám sát của một chuyên gia dinh dưỡng về chuyển hóa có kinh nghiệm và được xen kẽ với việc cho trẻ bú sữa công thức không chứa phenylalanine.
Sữa mẹ thường được giới hạn ở khoảng 25% số lần cho ăn, tùy thuộc vào mức độ nghiêm trọng của bệnh. Lượng phenylalanine hấp thụ qua sữa mẹ phải tính đến lượng phenylalanine hàng ngày. Sữa mẹ có hàm lượng phenylalanine thấp hơn so với sữa bột tiêu chuẩn dành cho trẻ sơ sinh (tương ứng là 14 mg/ounce so với 19 mg/ounce). Việc hạn chế chế độ ăn của mẹ không ảnh hưởng đến thành phần axit amin trong sữa mẹ.
Công thức dành cho trẻ lớn hơn và người lớn. Trẻ lớn hơn và người lớn tiếp tục uống hoặc ăn thực phẩm bổ sung dinh dưỡng không chứa phenylalanine (công thức tương đương protein), theo chỉ dẫn của nhà cung cấp dịch vụ chăm sóc sức khỏe hoặc chuyên gia dinh dưỡng. Liều lượng sữa công thức hàng ngày của bạn được chia thành các bữa ăn chính và ăn nhẹ, thay vì ăn hoặc uống tất cả cùng một lúc. Công thức dành cho trẻ lớn hơn và người lớn không giống như công thức dành cho trẻ sơ sinh, nhưng nó cũng cung cấp protein thiết yếu không chứa phenylalanine.
Tính ngon miệng kém của các chất thay thế protein ảnh hưởng xấu đến việc tuân thủ chế độ ăn kiêng, đặc biệt là ở trẻ lớn hơn. Lượng phenylalanine cần thiết được cung cấp với một lượng nhỏ protein tự nhiên. Hiếm bệnh nhân có thể cần bổ sung tyrosine.
Nên bắt đầu điều trị càng sớm càng tốt, thường là trước một tuần tuổi, ở trẻ sơ sinh có PKU và nồng độ phenylalanine trong máu >6 mg/dL (360 micromol/L). Việc điều trị nồng độ phenylalanine tăng cao nhưng thấp hơn một chút đang gây tranh cãi. Chúng tôi khuyên bạn nên điều trị cho trẻ sơ sinh có nồng độ phenylalanine dai dẳng từ 6 đến 10 mg/dL (360 đến 600 micromol/L). Thiếu BH4 nên được loại trừ là nguyên nhân của HPA.
Việc tiếp tục hạn chế chế độ ăn uống trong suốt cuộc đời dường như là cần thiết để có kết quả tối ưu.
Cơ quan Nghiên cứu Sức khỏe và Chất lượng đã tiến hành phân tích tổng hợp 17 nghiên cứu (bao gồm 432 cá nhân mắc PKU) kiểm tra mối quan hệ của phenylalanine trong máu với chỉ số IQ. Họ nhận thấy khả năng IQ thấp (<85) ngày càng tăng khi nồng độ phenylalanine trong máu cao hơn, bất kể IQ được đo trong thời thơ ấu hay sau đó, với mối liên hệ chặt chẽ hơn được thấy giữa phenylalanine được đo trong thời thơ ấu và IQ sau này.
Chế độ ăn uống suốt đời cũng được hỗ trợ bởi những phát hiện trong một nghiên cứu theo dõi dài hạn về trẻ sơ sinh mắc PKU tham gia thử nghiệm quản lý chế độ ăn uống. Trong thử nghiệm ban đầu, trẻ sơ sinh được điều trị bằng chế độ ăn hạn chế phenylalanine cho đến khi chúng được 6 tuổi và sau đó được chỉ định ngẫu nhiên để tiếp tục hoặc ngừng chế độ ăn kiêng. Vào khoảng 25 tuổi, 70 trong số 211 trẻ ban đầu được đánh giá lại. Trẻ em tiếp tục chế độ ăn kiêng giảm tỷ lệ mắc bệnh chàm (11 so với 28%), hen suyễn (0 so với 12%), nhức đầu (0 so với 31%), rối loạn tâm thần (22 so với 41%), hiếu động thái quá (0 so với 14%). và giảm hoạt động (0 so với 19 phần trăm) so với những người ngừng ăn kiêng. Việc tiếp tục chế độ ăn kiêng có liên quan đến điểm kiểm tra trí tuệ và thành tích tốt hơn cũng như nồng độ phenylalanine trong máu thấp hơn. Những bất thường trên MRI có liên quan đến nồng độ phenylalanine trong não cao hơn được đánh giá bằng quang phổ cộng hưởng từ.
Trong các nghiên cứu không ngẫu nhiên, điểm IQ vẫn ổn định ở trẻ lớn hơn và người lớn mắc PKU không ăn kiêng hạn chế. Tuy nhiên, sự thiếu sót tinh tế trong các biện pháp chú ý và tốc độ xử lý đã được ghi nhận khi so sánh với bệnh nhân ban đầu, được điều trị liên tục và/hoặc kiểm soát lành mạnh. Nồng độ phenylalanine cao đã được chứng minh là có ảnh hưởng đến tâm trạng và sự chú ý lâu dài ở người lớn trong một thử nghiệm chéo, ngẫu nhiên. Chín người lớn đã được bổ sung có chứa phenylalanine hoặc giả dược trong bốn tuần trong khi ăn kiêng hạn chế. Điểm số trên bảng câu hỏi Hồ sơ trạng thái tâm trạng (POMS) được điền bởi cả bệnh nhân và bạn bè hoặc người thân của họ thấp hơn đáng kể khi bổ sung phenylalanine so với giả dược.
Phác đồ điều trị bệnh Phenylketon niệu bằng thuốc
Liệu pháp dược lý chính có sẵn để điều trị PKU từ nhẹ đến trung bình là một công thức tổng hợp của BH4 được gọi là sapropterin.
BH4 là cofactor của PAH.
Pegylated phenylalanine amoniac lyase ( pegvaliase , PEG-PAL), một loại enzyme phân hủy phenylalanine, được phê duyệt là liệu pháp điều trị cho bệnh nhân trưởng thành mắc PKU.
* Tetrahydrobiopterin/sapropterin: Liều dược lý của BH4 là một biện pháp thay thế hoặc hỗ trợ cho việc hạn chế phenylalanine trong chế độ ăn ở những bệnh nhân mắc HPA hoặc kiểu hình PKU từ nhẹ đến trung bình. Phản ứng với BH4 ở trẻ em bị PKU (nhưng không thiếu BH4) đã được đánh giá trong một số nghiên cứu. Trong một trong những nghiên cứu lớn hơn, nồng độ phenylalanine trong máu được đo ở 557 trẻ sơ sinh và trẻ em mắc PKU sau khi dùng BH4 (20 mg/kg trọng lượng cơ thể). Với khả năng đáp ứng được xác định bằng việc giảm 30% nồng độ phenylalanine, 38% bệnh nhân phản ứng khi đo phenylalanine 8 giờ sau khi dùng và 46% phản hồi khi đo phenylalanine 24 giờ sau khi dùng. Tỷ lệ đáp ứng là 79 đến 83% ở những bệnh nhân mắc HPA (phenylalanine <10 mg/dL, 600 micromol/L), 49 đến 60% ở những bệnh nhân mắc PKU nhẹ (phenylalanine 10 đến 20 mg/dL, 600 đến 1200 micromol/ L) và 7 đến 10 phần trăm ở bệnh nhân PKU cổ điển (phenylalanine >20 mg/dL, 1200 micromol/L). Trong một nghiên cứu nhỏ hơn, khả năng đáp ứng với BH4 không được dự đoán nhất quán bởi kiểu gen.
* Sapropterin: một dạng tổng hợp có hoạt tính sinh học của BH4, đã được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) phê duyệt vào tháng 12 năm 2007. Sapropterin có thể được sử dụng như một chất hỗ trợ cho việc hạn chế chế độ ăn uống ở bệnh nhân PKU đáp ứng với sapropterin. Bệnh nhân nên được đề nghị dùng thử liệu pháp sapropterin để đánh giá khả năng đáp ứng, ngoại trừ những bệnh nhân có hai đột biến null trong trans. Khả năng đáp ứng của sapropterin thường được xác định bằng cách đạt được mức phenylalanine trong máu cơ bản vào ngày bắt đầu dùng thuốc và sau đó bắt đầu cho bệnh nhân dùng một liều sapropterin duy nhất hàng ngày ở mức 20 mg/kg. Nồng độ phenylalanine bổ sung trong máu sau đó được lấy theo các khoảng thời gian đều đặn, thường là sau 24 giờ, một tuần, hai tuần và trong một số trường hợp là ba hoặc bốn tuần.
Khả năng đáp ứng với sapropterin được xác định bằng cách sử dụng giao thức tải sapropterin thích hợp với thời gian quan sát ít nhất 24 giờ (48 giờ thì tốt hơn), sau đó là thử nghiệm sapropterin từ một đến bốn tuần với việc điều chỉnh liều lượng sapropterin và lượng phenylalanine sau đó để tối ưu hóa mức phenylalanine.
Ở trẻ em, liều khởi đầu được đề xuất là 10 mg/kg một lần mỗi ngày trong tối đa một tháng. Liều cuối cùng ở trẻ em và người lớn có thể được điều chỉnh trong khoảng từ 5 đến 20 mg/kg mỗi ngày, được chuẩn độ theo mức độ phenylalanine trong máu.
Trong một thử nghiệm đa trung tâm, 89 bệnh nhân mắc PKU được chỉ định ngẫu nhiên điều trị bằng sapropterin (10 mg/kg) hoặc giả dược trong sáu tuần. Liệu pháp sapropterin có liên quan đến việc giảm nồng độ phenylalanine trung bình trong máu là 3,9 mg/dL (236 micromol/L) so với mức tăng 0,05 mg/dL (3 micromol/L) ở nhóm giả dược; 44% bệnh nhân trong nhóm điều trị đã giảm ít nhất 30% nồng độ phenylalanine trong máu so với ban đầu so với 9% ở nhóm chứng. Chức năng thần kinh lâu dài ở bệnh nhân PKU được điều trị bằng sapropterin chưa được đánh giá.
Trong một nghiên cứu tiếp theo, 46 bệnh nhân (từ 4 đến 12 tuổi) đáp ứng với sapropterin được chỉ định ngẫu nhiên (3:1) dùng sapropterin (20 mg/kg mỗi ngày) hoặc giả dược trong 10 tuần. Các đối tượng tiếp tục thực hiện chế độ ăn kiêng hạn chế phenylalanine, nhưng sau ba tuần, các chất bổ sung phenylalanine trong chế độ ăn uống được thêm vào hoặc loại bỏ hai tuần một lần tùy thuộc vào việc kiểm soát phenylalanine. Khả năng dung nạp phenylalanine tăng lên ở những bệnh nhân được điều trị bằng sapropterin (chênh lệch trung bình đã điều chỉnh là 17,7 mg/kg mỗi ngày, KTC 95% là 9,0-27,0 mg/kg mỗi ngày).
* Phenylalanine amoniac lyase (PAL): PAL là một enzyme có nguồn gốc từ sinh vật nhân sơ Anabaena variabilis (Av) phân hủy phenylalanine. Một dạng (r) tái tổ hợp được kết hợp với polyethylen glycol (PEG) để giảm khả năng sinh miễn dịch (rAvPAL-PEG, pegvaliase ) đã được FDA Hoa Kỳ chấp thuận cho người lớn mắc PKU sau các thử nghiệm lâm sàng thành công chứng minh tính an toàn và hiệu quả lâu dài. PRISM-1 và PRISM-2, hai thử nghiệm lâm sàng giai đoạn III, đã đánh giá tính an toàn và hiệu quả của pegvaliase sau giai đoạn cảm ứng, chuẩn độ và duy trì. Bệnh nhân mắc PKU có nồng độ phenylalanine trong máu >600 micromol/L được chỉ định ngẫu nhiên theo tỷ lệ 1:1 với liều duy trì 20 mg/ngày hoặc pegvaliase 40 mg/ngày. Trong vòng 24 tháng điều trị, gần 70% bệnh nhân đạt được mức phenylalanine <600 micromol/L, với 60% đạt được mức <360 micromol/L. Các tác dụng phụ xảy ra thường xuyên hơn trong sáu tháng đầu điều trị. Các tác dụng phụ phổ biến nhất bao gồm đau khớp, phản ứng tại chỗ tiêm và đau đầu. Sốc phản vệ cấp tính rất hiếm (12/261 người tham gia). Trong một thử nghiệm giai đoạn I trước đó, tất cả bệnh nhân đã phát triển kháng thể chống lại PEG vào cuối nghiên cứu (ngày 42) và chưa đến một nửa đã phát triển kháng thể với PAL.
Các liệu pháp khác
* Axit béo không bão hòa đa chuỗi dài: Vì ít protein động vật nên chế độ ăn hạn chế phenylalanine dẫn đến nồng độ axit béo không bão hòa đa chuỗi dài (Long-Chain Polyunsaturated Fatty Acids, LCPUFA) và axit docosahexanoic (DHA) trong máu thấp, có thể ảnh hưởng đến sự phát triển thần kinh. Trong một thử nghiệm ngẫu nhiên ở trẻ em có HPA được kiểm soát tốt, việc bổ sung LCPUFA bao gồm DHA trong 12 tháng đã làm tăng nồng độ DHA trong máu và cải thiện chức năng thị giác so với giả dược [ 58 ] . Tuy nhiên, những giá trị này trở lại đường cơ sở sau ba năm [ 59 ]. Trong một thử nghiệm không ngẫu nhiên, việc bổ sung dầu cá (omega-3 LCPUFA) trong ba tháng đã cải thiện các kỹ năng vận động của trẻ có PKU được kiểm soát tốt so với nhóm đối chứng cùng lứa tuổi.
* Các axit amin trung tính lớn: LNAAs (arginine, histidine, isoleucine, leucine, lysine, methionine, threonine, tryptophan, tyrosine và valine) cạnh tranh với phenylalanine cho cùng một chất vận chuyển amino tại hàng rào máu não. Do đó, việc bổ sung LNAAs có thể làm giảm đáng kể dòng phenylalanine vào não ở bệnh nhân PKU. Trong một nghiên cứu, việc bổ sung LNAA (250 đến 500 mg/kg mỗi ngày) làm giảm nồng độ phenylalanine trong huyết tương do ức chế cạnh tranh hấp thu phenylalanine ở ruột non. LNAAs không được khuyến cáo cho trẻ nhỏ hoặc phụ nữ mang thai nhưng là một lựa chọn cho người lớn bị thiếu PAH, những người không kiểm soát chuyển hóa tốt và không tuân thủ các lựa chọn điều trị khác.
Bệnh Phenylketon niệu ở mẹ bầu (Phenylalanine Embryopathy)
Nồng độ phenylalanine huyết thanh tăng cao trong thời kỳ đầu mang thai ở người mẹ bị PKU hoặc chứng tăng phenylalanin máu (HPA) với nồng độ phenylalanine ổn định >360 micromol/L có thể dẫn đến bệnh phôi thai phenylalanine. Nguy cơ xuất phát từ việc kiểm soát trao đổi chất của người mẹ và không phụ thuộc vào việc thai nhi có PKU hay không. Phụ nữ bị PKU nên được khuyến khích nhận các dịch vụ kế hoạch hóa gia đình và tiền thụ thai. Con cái dường như không bị ảnh hưởng bởi chứng tăng phenylalanin máu nhẹ ở mẹ (MHPA), phenylalanine máu <360 micromol/L, một tình trạng khác với PKU.
Sinh bệnh học
Nồng độ phenylalanine trong thai nhi cao hơn trong huyết tương của mẹ. Trong một báo cáo, nồng độ trong máu của thai nhi thu được bằng phương pháp chọc dây rốn được so sánh với các mẫu tĩnh mạch của người mẹ đồng thời ở 14 trường hợp mang thai từ 19 tuần đến đủ tháng. Tỷ lệ nồng độ giữa thai nhi và mẹ tổng thể là 1,35 ± 0,42 (độ lệch chuẩn) và giảm dần trong khoảng thời gian đo. Do chênh lệch nồng độ, ngay cả khi nồng độ phenylalanine của mẹ nằm trong khoảng hợp lý, nồng độ của thai nhi có thể đạt đến mức gây quái thai.
Ảnh hưởng lâm sàng
Ảnh hưởng đối với phôi thai của PKU mẹ bao gồm hạn chế tăng trưởng trong tử cung, thiểu năng trí tuệ, tật đầu nhỏ và dị tật tim. Hình ảnh lâm sàng tương tự như bệnh phôi do rượu (alcohol embryopathy).
Nguy cơ bất thường phụ thuộc vào nồng độ phenylalanine trong máu của mẹ và không phụ thuộc vào kiểu gen của thai nhi (ví dụ: dị hợp tử hoặc đồng hợp tử đối với PKU). Ở những trường hợp mang thai không được điều trị mà nồng độ phenylalanine trong máu của người mẹ ≥20 mg/dL (1200 micromol/L), tật đầu nhỏ và thiểu năng trí tuệ xảy ra ở 73 đến 92% trẻ sơ sinh và 12% mắc bệnh tim bẩm sinh.
Kết quả được cải thiện đáng kể khi việc điều trị dẫn đến nồng độ phenylalanine ở người mẹ thấp một cách lý tưởng trước khi thụ thai hoặc ít tối ưu hơn trước khi thai được 10 tuần. Điều này được minh họa bởi Nghiên cứu PKU dành cho bà mẹ, một dự án hợp tác quốc tế đánh giá tiền cứu 572 trường hợp mang thai ở phụ nữ có PKU và 99 đối chứng. Trong số những bà mẹ bị ảnh hưởng đã sinh con sống, kiểm soát trao đổi chất đã đạt được trước khi thụ thai hoặc 10 tuần với tỷ lệ tương ứng là 16 và 18%. Ở những nhóm này, tỷ lệ tật đầu nhỏ lần lượt là 3,6% và 5%, thấp hơn đáng kể so với tỷ lệ ở những thai kỳ không được điều trị.
Nguy cơ mắc bệnh tim bẩm sinh ở con cái phụ thuộc vào nồng độ phenylalanine trong máu của mẹ lúc ban đầu và trong giai đoạn hình thành tim (4 đến 10 tuần tuổi thai). Trong một báo cáo khác từ Nghiên cứu PKU dành cho bà mẹ được trích dẫn ở trên, con cái được so sánh từ các trường hợp mang thai bị ảnh hưởng và kiểm soát. Bệnh tim bẩm sinh xảy ra ở 34 trong số 235 đứa con (14%) của những bà mẹ có mức phenylalanine cơ bản ≥15 mg/dL (≥900 micromol/L) và mức dai dẳng ≥10 mg/dL (≥600 micromol/L) theo thời gian. tuần thứ tám của thai kỳ so với 1 trong số 99 đối chứng (1%). Tỷ lệ hẹp động mạch chủ (20%) và hội chứng giảm sản tim trái (11%) cao hơn dự kiến trong dân số nói chung.
Một báo cáo khác từ Nghiên cứu PKU dành cho bà mẹ đã xem xét ảnh hưởng của tật đầu nhỏ và bệnh tim bẩm sinh ở trẻ đối với kết quả phát triển được đo bằng Chỉ số Nhận thức Tổng quát McCarthy khi trẻ bốn tuổi và Thang đo Trí thông minh Wechsler cho Trẻ em Sửa đổi khi trẻ sáu tuổi. Tỷ lệ chung của bệnh tim bẩm sinh và tật đầu nhỏ lần lượt là 7,7% và 33%. Trẻ sơ sinh mắc cả hai dị tật có nồng độ phenylalanine ở mẹ cao hơn so với những trẻ chỉ mắc bệnh tim. Tất cả trẻ sơ sinh mắc bệnh tim đều có nồng độ phenylalanine ở mẹ trên 6 mg/dL (360 micromol/L trong 8 đến 10 tuần đầu tiên của thai kỳ). So với những đứa trẻ không có bất thường nào, chỉ số thông minh (IQ) thấp hơn với bệnh đầu nhỏ hoặc bệnh tim bẩm sinh và thậm chí thấp hơn với cả hai. Giảm chỉ số IQ có liên quan đến việc tăng phơi nhiễm phenylalanine.
Các phát hiện tương tự như kết quả của Nghiên cứu PKU dành cho bà mẹ đã được mô tả trong một báo cáo hồi cứu từ Cơ quan đăng ký PKU của Vương quốc Anh từ năm 1978 đến 1997. Trong báo cáo này, những đứa trẻ được sinh ra từ những bà mẹ bắt đầu chế độ ăn kiêng hạn chế phenylalanine trước khi thụ thai có kết quả sau so với những đứa trẻ có mẹ bắt đầu chế độ ăn kiêng trong khi mang thai:
- Cân nặng khi sinh trung bình lớn hơn (3160 so với 2818 gram)
- Vòng đầu khi sinh trung bình lớn hơn (33,6 so với 32,7 cm)
- Chỉ số phát triển trung bình (DQ) lớn hơn sau 4 năm (108,9 so với 96,8)
- DQ trung bình cao hơn sau 8 năm (103,4 so với 86,5)
- Tỷ lệ mắc bệnh tim bẩm sinh thấp hơn (2,4% so với 17%)
Phòng ngừa PKU ở phụ nữ mang thai
Bệnh phôi phenylalanine có thể được ngăn ngừa bằng cách hạn chế lượng phenylalanine trong chế độ ăn uống ở phụ nữ bị PKU hoặc HPA trước và trong khi mang thai.
Tuyên bố Phát triển Đồng thuận của Viện Y tế Quốc gia (NIH) khuyến nghị rằng nên giảm nồng độ phenylalanine trong huyết tương xuống mức <6 mg/dL (360 micromol/L) ít nhất ba tháng trước khi thụ thai và duy trì ở mức 2 đến 6 mg/dL (120 đến 360 micromol/L) khi mang thai.
Những bà mẹ thụ thai trong khi nồng độ trong máu cao hơn mức khuyến nghị nên đạt được sự kiểm soát trao đổi chất càng sớm càng tốt. Nồng độ phenylalanine trong huyết tương nên được theo dõi hai lần một tuần hoặc tối thiểu một lần một tuần. Lượng phenylalanine dung nạp tăng lên trong thai kỳ. Chế độ ăn uống nên được điều chỉnh theo mức huyết tương.
Nồng độ tyrosine trong huyết tương của người mẹ nên được duy trì trong khoảng từ 0,9 đến 1,8 mg/dL. Có thể cần bổ sung tyrosine để duy trì ngưỡng nồng độ này.
Tài liệu tham khảo
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010; 376:1417.
- van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017; 12:162.
- Blau N. Genetics of Phenylketonuria: Then and Now. Hum Mutat 2016; 37:508.
- Flydal MI, Martinez A. Phenylalanine hydroxylase: function, structure, and regulation. IUBMB Life 2013; 65:341.
- MacDonald A. Diet and compliance in phenylketonuria. Eur J Pediatr 2000; 159 Suppl 2:S136.
- Yi SH, Singh RH. Protein substitute for children and adults with phenylketonuria. Cochrane Database Syst Rev 2015; :CD004731.
- Clemens PC, Heddrich-Ellerbrok M, Wachtel V, Link RM. Plasma amino acids in adolescents and adults with phenylketonuria on three different levels of protein intake. Acta Paediatr Scand 1991; 80:577.
- Webster D, Wildgoose J. Tyrosine supplementation for phenylketonuria. Cochrane Database Syst Rev 2013; :CD001507.
- Maternal phenylketonuria. Pediatrics 2008; 122:445.
(*) Theo UpToDate