Bệnh Alpha thalassemia là một thuật ngữ chung cho một nhóm các rối loạn máu di truyền được đặc trưng bởi việc giảm hoặc không sản xuất các tiểu đơn vị alpha-globin, dẫn đến nồng độ huyết sắc tố thấp.
Nội dung:
Tổng quan chung về bệnh Alpha Thalassemia
Alpha thalassemia là một thuật ngữ chung cho một nhóm các rối loạn máu di truyền được đặc trưng bởi việc giảm hoặc không sản xuất các tiểu đơn vị alpha-globin, dẫn đến nồng độ huyết sắc tố thấp và không có đầy đủ chức năng.
Huyết sắc tố (Hemoglobin) được tìm thấy trong các tế bào hồng cầu; đó là sắc tố màu đỏ, giàu sắt, mang oxy của máu. Chức năng chính của các tế bào hồng cầu là cung cấp oxy đi khắp cơ thể.
Có hai dạng chính của bệnh alpha thalassemia có liên quan đến các vấn đề sức khỏe nghiêm trọng:
- bệnh phù thai do huyết sắc tố (Hb) Bart
- bệnh huyết sắc tố H (HbH)
Bệnh phù thai do huyết sắc tố (Hb) Bart là một hội chứng nghiêm trọng thường gây tử vong cho phôi thai đang phát triển trong thời kỳ mang thai hoặc ngay sau khi sinh; tuy nhiên, những tiến bộ gần đây đã dẫn đến các phương pháp điều trị được cải thiện cho tình trạng này.
Bệnh HbH rất đa dạng, và các triệu chứng cụ thể và mức độ nghiêm trọng có thể khác nhau rất nhiều từ người này sang người khác. Một số cá nhân sẽ chỉ có các triệu chứng nhỏ, trong khi những người khác sẽ phát triển các biến chứng nghiêm trọng tiềm ẩn.
Dấu hiệu đặc trưng của tất cả các dạng bệnh alpha thalassemia là thiếu máu, các tế bào hồng cầu nhỏ (hồng cầu nhỏ), chứa hàm lượng huyết sắc tố chức năng thấp (giảm sắc tố), và có thể bị phân hủy sớm ở cả tủy xương (tạo hồng cầu không hiệu quả) và trong tuần hoàn ngoại vi (tan máu).
Do đó, những người bị ảnh hưởng nghiêm trọng có thể không lưu thông đủ máu giàu oxy khắp cơ thể. Những cá nhân này có thể cảm thấy mệt mỏi, suy nhược, khó thở, chóng mặt hoặc đau đầu.
Thiếu máu nặng có thể gây ra các biến chứng nghiêm trọng, thậm chí đe dọa tính mạng nếu không được điều trị. Những người mắc bệnh HbH dạng nặng thường được điều trị bằng truyền máu thường xuyên, điều này có thể dẫn đến tích tụ sắt dư thừa trong cơ thể (thừa sắt).
Mặc dù tình trạng quá tải sắt có thể gây tổn thương nhiều cơ quan trong cơ thể, nhưng tình trạng này có thể được điều trị hiệu quả bằng một số loại thuốc có hiệu quả cao.
Đặc điểm di truyền của Alpha thalassemia
Điều quan trọng cần nhớ là không có “gen alpha thalassemia” gây bệnh. Tất cả chúng ta đều có gen alpha globin có liên quan đến căn bệnh này. Một số người chỉ có phiên bản gen này bị hư hỏng và đây chính là nguyên nhân gây ra bệnh.
Điều khiến bệnh alpha thalassemia trở nên phức tạp là do có hai gen alpha globin là HBA1 và HBA2. Điều này có nghĩa là mọi người đều có 4 bản sao gen alpha globin.
Mỗi bản sao của một gen được gọi là một alen và một người nhận một alen từ cha và một alen từ mẹ. Bởi vì có hai gen liên quan nên bốn alen phối hợp với nhau để tạo ra protein alpha globin.
Hầu hết các cá nhân thừa hưởng hai bản sao của mỗi gen (tổng cộng là 4 gen); một trong số mỗi gen từ cha của một người và một trong số mỗi gen từ mẹ của một người.
- Một đột biến ở bất kỳ một trong 4 gen alpha dẫn đến tình trạng không có triệu chứng (người mang mầm bệnh alpha thalassemia thầm lặng), nhưng các cá nhân có thể truyền gen đột biến cho con cái của họ.
- Một đột biến (hoặc nhiều đột biến) ảnh hưởng đến 2 trong số 4 gen alpha dẫn đến tình trạng không có triệu chứng hoặc chỉ có các triệu chứng rất nhẹ (alpha thalassemia thể nhẹ).
- Một đột biến (hoặc nhiều đột biến) ảnh hưởng đến 3 gen alpha dẫn đến bệnh HbH, trong khi các khiếm khuyết ảnh hưởng đến cả 4 gen dẫn đến bệnh phù thai nhi Hb Bart.
Đặc điểm triệu chứng của người mắc bệnh Alpha Thalassemia
Các triệu chứng cụ thể và mức độ nghiêm trọng của tình trạng bệnh alpha thalassemia rất khác nhau giữa người này với người khác.
Những người mang mầm bệnh alpha thalassemia thầm lặng không phát triển các triệu chứng, trong khi những người mắc bệnh alpha thalassemia thể nhẹ không phát triển bất kỳ triệu chứng nào hoặc chỉ bị thiếu máu nhẹ.
Nhiều người mắc một trong hai dạng bệnh alpha thalassemia trải qua cuộc đời mà không bao giờ biết rằng họ mang (những) gen biến đổi của chứng rối loạn này.
Trong một số trường hợp, chẩn đoán được thực hiện tình cờ trong khi họ đang được đánh giá về một tình trạng khác.
Hai dạng bệnh alpha thalassemia có liên quan đến các triệu chứng quan trọng, bệnh huyết sắc tố H và bệnh phù thai do Hb Bart.
BỆNH HUYẾT SẮC TỐ H (HbH)
Các triệu chứng cụ thể và mức độ nghiêm trọng của bệnh HbH có thể khác nhau rất nhiều từ người này sang người khác.
Một số cá nhân không phát triển các triệu chứng và chỉ nhận thức được rối loạn khi xét nghiệm máu định kỳ. Trong một số trường hợp, những người bị ảnh hưởng không phát triển các triệu chứng cho đến tuổi trưởng thành.
Hầu hết các cá nhân biểu hiện các triệu chứng liên quan đến thiếu máu nhẹ đến trung bình. Tuy nhiên, một số cá nhân sẽ phát triển các triệu chứng nghiêm trọng có thể phát triển trong thời thơ ấu hoặc thậm chí trong năm đầu tiên của cuộc đời.
Điều quan trọng cần lưu ý là những người bị ảnh hưởng có thể không có tất cả các triệu chứng được thảo luận bên dưới. Các cá nhân bị ảnh hưởng hoặc cha mẹ của trẻ em bị ảnh hưởng nên nói chuyện với bác sĩ và chuyên gia y tế về trường hợp cụ thể của họ, các triệu chứng liên quan và tiên lượng tổng thể.
Mức độ nghiêm trọng của bệnh bị ảnh hưởng một phần bởi loại đột biến cụ thể hiện diện.
Bệnh HbH có thể được gây ra bởi các đột biến xóa hoặc không xóa, đơn độc hoặc kết hợp.
- Bệnh HbH mất đoạn xảy ra khi sự kết hợp của các đột biến mất đoạn loại bỏ ba trong số bốn gen biểu hiện protein alpha-globin. Đây là dạng bệnh HbH phổ biến nhất.
- Bệnh HbH không xóa gen xảy ra khi một đột biến xóa loại bỏ hai gen alpha và một đột biến điểm không xóa gen làm bất hoạt gen thứ ba mà không loại bỏ nó một cách vật lý.
- Các đột biến không mất đoạn thường liên quan đến tình trạng thiếu máu trầm trọng hơn, có nhiều khả năng gây phì đại lá lách và gan, đồng thời có nhiều khả năng cần truyền máu điều trị hơn.
Bệnh HbH thường có biểu hiện thiếu máu, có thể ở các mức độ và mức độ nghiêm trọng khác nhau.
- Thiếu máu có thể dẫn đến mệt mỏi, suy nhược, khó thở, choáng váng, nhức đầu và vàng da, niêm mạc và lòng trắng mắt (vàng da).
- Trẻ sơ sinh bị ảnh hưởng nghiêm trọng thường không phát triển và tăng cân như mong đợi dựa trên độ tuổi và giới tính (không phát triển mạnh). Một số trẻ sơ sinh trở nên nhợt nhạt dần (xanh xao).
- Các vấn đề về ăn uống, khó chịu hoặc quấy khóc, gan to bất thường (gan to) và lách to bất thường (lách to) cũng có thể xảy ra.
- Thiếu tăng trưởng có thể xảy ra trong một số trường hợp.
Lách to có thể gây phì đại hoặc sưng bụng.
- Lách to có thể liên quan đến lá lách hoạt động quá mức (cường lách), một tình trạng có thể phát triển do quá nhiều tế bào máu tích tụ và bị phá hủy trong lá lách.
- Cường lách có thể góp phần gây thiếu máu ở những người mắc bệnh alpha thalassemia và gây ra lượng bạch cầu thấp, làm tăng nguy cơ nhiễm trùng và lượng tiểu cầu thấp, có thể dẫn đến chảy máu.
Các triệu chứng khác có thể xảy ra bao gồm các khối hình thành do sản xuất tế bào máu bên ngoài tủy xương (tạo máu ngoài tủy). Những khối này chủ yếu hình thành ở lá lách, gan, ngực và cột sống. Những khối này có khả năng gây chèn ép các cấu trúc lân cận và gây ra nhiều triệu chứng khác nhau.
Những người bị ảnh hưởng cũng có thể biểu hiện loét chân, sỏi mật và thiếu axit folic. Ngoài ra, bệnh HbH có xu hướng trở nên tồi tệ hơn khi các cá nhân dùng thuốc oxy hóa, tiếp xúc với một số hóa chất hoặc bị nhiễm trùng do tốc độ phá hủy hồng cầu (tan máu) tăng lên.
Một số người lớn tuổi mắc bệnh HbH, cũng như những người được điều trị bằng cách truyền máu thường xuyên, có thể bị quá tải sắt, một tình trạng đặc trưng bởi sự tích tụ sắt trong các mô khác nhau của cơ thể.
Tình trạng ứ sắt có thể gây tổn thương mô và suy giảm chức năng của các cơ quan bị ảnh hưởng như tim, gan và các tuyến nội tiết.
Tình trạng quá tải sắt có thể làm hỏng tim và gây ra nhịp tim bất thường, viêm màng ngoài tim (màng ngoài tim) lót tim (viêm màng ngoài tim), mở rộng tim và bệnh cơ tim (bệnh cơ tim giãn nở).
- Sự tham gia của tim cuối cùng có thể tiến triển thành các biến chứng đe dọa tính mạng như suy tim.
- Sự tham gia của gan có thể gây ra sẹo và viêm gan (xơ gan) và huyết áp cao của tĩnh mạch chính của gan (tăng áp lực tĩnh mạch cửa).
- Sự tham gia của các tuyến nội tiết có thể gây ra sự thiếu hụt của một số tuyến như tuyến giáp (suy giáp) và tuyến tụy (đái tháo đường).
Tình trạng quá tải sắt là một biến chứng của việc truyền máu nhiều lần có thể được sử dụng để điều trị cho một số người mắc bệnh HbH. Tuy nhiên, nhiều người lớn chưa bao giờ được truyền máu đã bị quá tải sắt, rất có thể là do tăng hấp thu sắt từ đường tiêu hóa.
Hemoglobin H-Constant Spring
Đây là một dạng biến thể của bệnh HbH và là dạng rối loạn không mất protein phổ biến nhất.
Những người mắc bệnh huyết sắc tố H-Constant Spring có xu hướng bị thiếu máu trầm trọng hơn vì quá trình sản xuất hồng cầu thậm chí còn kém hiệu quả hơn so với các dạng bệnh HbH không tiêu hủy (tạo hồng cầu không hiệu quả).
Lách to vừa phải là phổ biến ở những người này.
Các triệu chứng phổ biến khác bao gồm loét chân, sỏi mật, vàng da và tăng nguy cơ nhiễm trùng. Sự chậm phát triển có ý nghĩa hơn ở trẻ em bị ảnh hưởng so với trẻ em mắc bệnh HbH.
Những người bị ảnh hưởng có thể có nguy cơ đặc biệt bị thiếu máu nghiêm trọng, đột ngột phát triển sau một đợt sốt cấp tính, đây là thuật ngữ không đặc hiệu cho bất kỳ bệnh nào, mặc dù bệnh này thường khởi phát nhanh, kèm theo sốt.
BỆNH PHÙ THAI DO HB BART
Thai nhi bị phù nước Hb Bart, còn được gọi là bệnh alpha thalassemia thể nặng, là dạng bệnh alpha thalassemia nghiêm trọng nhất.
Thuật ngữ thai nhi phù nề mô tả sự tích tụ một lượng lớn chất lỏng (phù nề) trong các mô và cơ quan của thai nhi đang phát triển. Phù lan rộng (diffuse).
Thai nhi đang phát triển cũng có thể biểu hiện thiếu máu trầm trọng, gan to bất thường (gan to), lá lách to bất thường (lách to), não phát triển kém và các dấu hiệu suy tim.
Sự tích tụ bất thường của dịch não tủy trong hộp sọ (não úng thủy) cũng có thể xảy ra. Não úng thủy gây tăng áp lực và sưng não. Trẻ sơ sinh có thể nhợt nhạt và biểu hiện những bất thường về xương và đường tiết niệu (niệu sinh dục).
Bệnh phù thai do Hb Bart thường gây tử vong trước khi sinh (thai chết lưu) hoặc ngay sau khi sinh (thời kỳ sơ sinh).
Nguyên nhân gây ra Alpha Thalassemia
Alpha thalassemia gây ra bởi sự thay đổi (đột biến) ở hai gen liền kề, gen HBA1 và HBA2.
Mỗi người có hai bản sao của gen HBA1 (một từ cha hoặc mẹ) và hai bản sao của gen HBA2 (cũng là một từ cha và mẹ).
Các cá nhân bị ảnh hưởng có thể có một đột biến hoặc tổ hợp các đột biến ở một gen, hai gen, ba gen hoặc cả bốn bản sao của những gen này.
Các gen alpha mang thông tin di truyền mã hóa quá trình tổng hợp tạo ra các protein đóng vai trò quan trọng trong nhiều chức năng của cơ thể.
Khi xảy ra đột biến gen, sản phẩm protein có thể hoạt động bình thường nhưng bị giảm về số lượng, hoặc hoạt động không bình thường và được tạo ra ở mức bình thường.
Tùy thuộc vào chức năng của protein, điều này có thể ảnh hưởng đến nhiều hệ thống cơ quan của cơ thể.
Các bệnh di truyền được xác định bởi sự kết hợp của các gen cho một đặc điểm cụ thể nằm trên nhiễm sắc thể nhận được từ cha và mẹ.
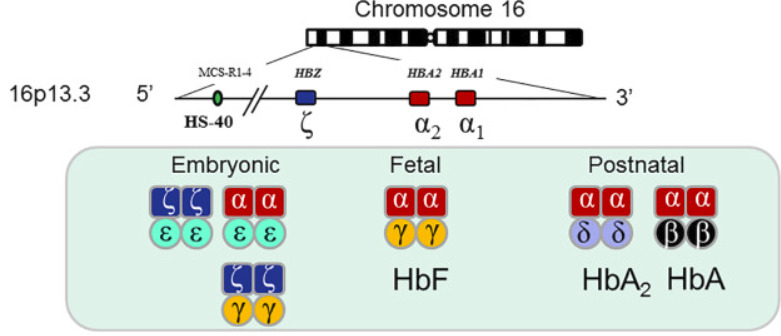
Các nhà khoa học đã xác định rằng gen HBA1 và HBA2 nằm trên nhánh ngắn (p) của nhiễm sắc thể 16 (16p13.3).
Nhiễm sắc thể, hiện diện trong nhân tế bào người, mang thông tin di truyền cho mỗi cá nhân. Tế bào người bình thường có 46 nhiễm sắc thể. Các cặp nhiễm sắc thể của con người được đánh số từ 1 đến 22 và nhiễm sắc thể giới tính được ký hiệu là X và Y.
Nam giới có một nhiễm sắc thể X và một nhiễm sắc thể Y và nữ giới có hai nhiễm sắc thể X.
Mỗi nhiễm sắc thể có một nhánh ngắn được ký hiệu là “p” và một nhánh dài được ký hiệu là “q”. Các nhiễm sắc thể được chia nhỏ hơn nữa thành nhiều dải được đánh số.
Các gen HBA1 và HBA2 quy định việc tổng hợp chuỗi protein alpha globin.
Có ba loại huyết sắc tố chính: phôi thai, thai nhi và người lớn.
- Hemoglobin phôi thai được tạo ra trong vài tháng đầu tiên sau khi thụ thai. Các huyết sắc tố của thai nhi bắt đầu biểu hiện khi thai được tám tuần và nhanh chóng thay thế các huyết sắc tố của phôi thai.
- Bắt đầu từ khi sinh ra, huyết sắc tố bào thai được thay thế bằng huyết sắc tố trưởng thành trong một quá trình mà phần lớn được hoàn thành ở độ tuổi 6-12 tháng.
- Huyết sắc tố bình thường được tạo thành từ các protein chuyên biệt gọi là globin.
Hemoglobin bào thai và người trưởng thành bao gồm hai chuỗi alpha và hai chuỗi protein khác, hoặc là chuỗi gamma (trong huyết sắc tố bào thai) hoặc chuỗi beta (trong huyết sắc tố trưởng thành).
Các thay đổi trong gen mã hóa Alpha Thalassemia chủ yếu là do mất đi các đoạn có độ dài hoặc đột biến điểm khác nhau.
Đột biến điểm là sự thay thế một bazơ hoặc thêm hoặc xóa một lượng nhỏ.
Những loại đột biến khác nhau này có thể được phát hiện bằng phương pháp PCR đặc hiệu với alen, phân tích vết chấm ngược (RDB), Gap-PCR, PCR thời gian thực với phân tích đường cong nóng chảy và giải trình tự ADN. Trình tự mồi được công bố để chẩn đoán một số đột biến xóa α+ hoặc α°.
* Đột biến ở một gen alpha dẫn đến việc sản xuất chuỗi alpha chức năng thấp hơn một chút và không gây ra bất kỳ triệu chứng nào (người mang mầm bệnh thalassemia alpha thầm lặng).
* Đột biến ở hai gen làm giảm sản xuất chuỗi alpha chức năng, nhưng không đủ để gây ra các triệu chứng đáng kể, mặc dù một số người có thể bị thiếu máu nhẹ (alpha thalassemia thể nhẹ). Khi hai gen đột biến nằm trên cùng một nhiễm sắc thể 16, nó được gọi là xóa đoạn ‘cis’; khi một gen đột biến từ một nhiễm sắc thể 16 và gen đột biến khác từ nhiễm sắc thể 16 khác, nó được gọi là xóa ‘trans’.
* Một đột biến ở ba gen dẫn đến giảm đáng kể quá trình sản xuất chuỗi alpha (bệnh huyết sắc tố H). Việc giảm hoặc thiếu chuỗi protein alpha dẫn đến mất cân bằng với chuỗi protein beta được biểu hiện ở số lượng bình thường. Khi các chuỗi beta hiện diện quá nhiều (như xảy ra trong bệnh Hb H), các chuỗi dư thừa này sẽ liên kết với nhau để tạo ra một loại huyết sắc tố bất thường được gọi là huyết sắc tố H. Huyết sắc tố H không ổn định và khiến các tế bào hồng cầu bị phá vỡ nhanh hơn bình thường trong tủy xương (tạo hồng cầu không hiệu quả) và trong tuần hoàn ngoại biên (tan máu). Hemoglobin H-Constant Spring là một dạng bệnh HbH bất thường được đặc trưng bởi một quá trình lâm sàng tồi tệ hơn đáng kể và khác với các dạng bệnh Hb H phổ biến hơn ở chỗ một (trong ba) gen alpha bị ảnh hưởng mang đột biến không xóa.
* Một đột biến ở cả bốn gen dẫn đến giảm nghiêm trọng hoặc không sản xuất chuỗi alpha (bệnh phù thai do Hb Bart).
Đột biến trong gen alpha được di truyền theo gen lặn trên nhiễm sắc thể thường.
Các rối loạn di truyền lặn trở nên biểu hiện khi một cá thể thừa hưởng một đột biến của gen tương ứng từ cả bố và mẹ.
Nếu một người nhận được một gen bình thường và một gen bệnh thì người đó sẽ là người mang mầm bệnh nhưng thường không biểu hiện triệu chứng.
Do đó:
- Nguy cơ cả bố và mẹ mang gen đột biến Alpha thalassemia đều truyền một gen khiếm khuyết cho con cái và sinh ra một đứa trẻ bị bệnh Thalassemia là 25% cho mỗi lần mang thai.
- Nguy cơ sinh con cũng là người mang mầm bệnh (giống như cha mẹ) là 50% cho mỗi lần mang thai.
- Cơ hội để một đứa trẻ nhận được gen bình thường từ cả bố và mẹ và bình thường về mặt di truyền đối với đặc điểm cụ thể đó là 25% cho mỗi lần mang thai.
Đối với các gen mang trên nhiễm sắc thể 1-22, nguy cơ mắc bệnh là như nhau đối với nam và nữ. Sự di truyền của bệnh alpha thalassemia có phần phức tạp hơn khi mỗi cha mẹ đóng góp hai gen alpha cho con cái, thay vì chỉ một.
Các gen HBA1 và HBA2 được di truyền theo cặp, nghĩa là cả hai gen từ một nhiễm sắc thể được truyền từ cha mẹ sang con cái.
Nên tham khảo ý kiến của chuyên gia tư vấn di truyền đối với các gia đình hoặc cha mẹ đã biết hoặc nghi ngờ mang đột biến alpha thalassemia, ngay cả khi nó không gây ra triệu chứng.
Các nhà nghiên cứu đã xác định rằng sự tiến triển và mức độ nghiêm trọng của bệnh alpha thalassemia có xu hướng thay đổi dựa trên loại đột biến cụ thể có trong (các) gen cũng như vị trí cụ thể của đột biến trên (các) gen đó. Điều này được gọi là tương quan kiểu gen-kiểu hình và cho phép các bác sĩ dự đoán những cá nhân có nguy cơ phát triển các triệu chứng nghiêm trọng hơn (ví dụ: những người có HbH-Constant Spring). Tuy nhiên, do số lượng gen liên quan, sự biểu hiện của kiểu gen và kiểu hình trong bệnh alpha thalassemia rất đa dạng và khác nhau, và mối tương quan giữa kiểu gen và kiểu hình cụ thể vẫn chưa được hiểu rõ hoàn toàn. Cần nhiều nghiên cứu hơn để làm rõ đầy đủ mối tương quan giữa kiểu gen và kiểu hình trong bệnh alpha thalassemia.
Các nhà nghiên cứu cũng tin rằng các yếu tố bổ sung ảnh hưởng đến mức độ nghiêm trọng của bệnh HbH và Hb Bart’s hydrops thai nhi, bao gồm các gen điều chỉnh và các yếu tố môi trường. Gen điều chỉnh, không giống như gen gây bệnh alpha thalassemia, có thể ảnh hưởng đến mức độ nghiêm trọng lâm sàng của chứng rối loạn. Cần có nhiều nghiên cứu hơn để khám phá các yếu tố di truyền và môi trường khác nhau liên quan đến bệnh alpha thalassemia và vai trò chính xác của chúng trong sự phát triển của rối loạn.
Sự phân bố của bệnh Alpha Thalassemia
Alpha thalassemia là một trong những rối loạn di truyền lặn trên nhiễm sắc thể thường phổ biến nhất trên thế giới với tần số gen thay đổi từ 1% đến 98% trên khắp vùng nhiệt đới và cận nhiệt đới.
Không giống như Beta-thalassemia, trong đó các đột biến không mất đoạn chiếm ưu thế, > 95% các trường hợp bệnh Alpha thalassemia được công nhận liên quan đến việc xóa 1 hoặc cả hai gen α-globin khỏi nhiễm sắc thể 16p13.3.
Phổ biến nhất trong số các biến thể đột biến của Alpha thalassemia là:
- -α. 3,7 và -α. 4.2 xóa bỏ gen đơn
- – SEA và – FIL Đông Nam Á xóa gen kép
- – MED và -α. 20.5 Địa Trung Hải xóa gen kép
Alpha thalassemia được tìm thấy ở hầu hết các quần thể trên toàn thế giới, nhưng phổ biến nhất ở Trung Đông, Đông Nam Á và một số quốc gia Địa Trung Hải. Bệnh phù thai do Hb Bart và bệnh HbH chủ yếu được công nhận ở Đông Nam Á. Tỷ lệ ước tính của bệnh phù thai do Hb Bart ở Đông Nam Á là 1 trên 200-2.000 ca sinh; tỷ lệ mắc bệnh ở các nơi khác trên thế giới là không rõ. Tỷ lệ mắc bệnh HbH ở các quốc gia này là khoảng 4-20 người trên 1.000 ca sinh.
Sự gia tăng nhập cư của những người từ các khu vực có tỷ lệ mắc bệnh Alpha thalassemia cao hơn đã dẫn đến tỷ lệ mắc bệnh rối loạn alpha-globin tăng lên ở Hoa Kỳ và các quốc gia phương Tây khác. Mặc dù tỷ lệ mắc bệnh và tỷ lệ lưu hành đang gia tăng ở Hoa Kỳ và Bắc Âu, tỷ lệ mắc bệnh hoặc tỷ lệ lưu hành chính xác vẫn chưa được biết. Các thể nặng của bệnh alpha thalassemia (bệnh HbH và thai nhi bị phù nước Hb Bart) đã được ước tính xảy ra ở khoảng 1 trên 1.000.000 cá nhân trong dân số nói chung ở Bắc Âu và Bắc Mỹ. Tuy nhiên, một số nghiên cứu đã chỉ ra rằng bệnh alpha thalassemia có thể chưa được công nhận và chẩn đoán chưa đầy đủ ở những quốc gia này, gây khó khăn cho việc xác định tần suất thực sự của bệnh.
Một số nghiên cứu đã ước tính rằng có tới 5% dân số thế giới mang biến thể alpha-thalassemia (nghĩa là đột biến ở một trong hai cặp gen liên quan đến bệnh alpha thalassemia).
Chẩn đoán bệnh Alpha Thalassemia
Chẩn đoán bệnh alpha thalassemia dựa trên việc xác định các triệu chứng đặc trưng, bệnh sử chi tiết của bệnh nhân, đánh giá lâm sàng kỹ lưỡng và nhiều xét nghiệm chuyên biệt. Hb Bart’s hydrops thai nhi có thể được chẩn đoán trước khi sinh trong hầu hết các trường hợp.
Tại Hoa Kỳ, trẻ sơ sinh có thể được chẩn đoán mắc bệnh alpha thalassemia thông qua sàng lọc sơ sinh.
Sàng lọc sơ sinh là một chương trình y tế công cộng quy định việc đánh giá trẻ sơ sinh về nhiều loại rối loạn có thể điều trị được nhưng không biểu hiện rõ ràng khi sinh. Chương trình sàng lọc trẻ sơ sinh của mỗi tiểu bang (và các rối loạn cụ thể được thử nghiệm) là khác nhau. Cần thử nghiệm thêm để xác định chính xác loại bệnh alpha thalassemia hiện có.
Xét Nghiệm Lâm Sàng và Kiểm Tra
Các Bác Sĩ sẽ lấy mẫu máu từ những người bị nghi ngờ mắc một trong các bệnh alpha thalassemia. Một số xét nghiệm khác nhau có thể được thực hiện trên một mẫu máu.
Những người bị nghi ngờ mắc bệnh alpha thalassemia sẽ trải qua các xét nghiệm máu như xét nghiệm công thức máu toàn phần (CBC). CBC đo lường một số thành phần và khía cạnh của máu bao gồm số lượng, nồng độ, kích thước, hình dạng và sự trưởng thành của các tế bào máu.
Một xét nghiệm máu chuyên biệt được gọi là điện di huyết sắc tố đo các loại huyết sắc tố khác nhau được tìm thấy trong máu.
Với bệnh alpha thalassemia, CBC là cần thiết để đo lượng huyết sắc tố và số lượng cũng như kích thước và hình dạng của các tế bào hồng cầu, những tế bào này có số lượng ít hơn và kích thước nhỏ hơn (hồng cầu nhỏ) so với những người không mắc bệnh alpha thalassemia. Các tế bào hồng cầu cũng có thể có màu nhạt (hypochromic) và có nhiều hình dạng khác nhau. Một mẫu máu cũng có thể được xét nghiệm để đo lượng sắt trong máu, có thể tăng cao ở một số người mắc bệnh alpha thalassemia.
Xét nghiệm di truyền phân tử có thể xác nhận chẩn đoán bệnh alpha thalassemia. Xét nghiệm di truyền phân tử có thể phát hiện các đột biến trong gen HBA1 và HBA2 được biết là nguyên nhân gây ra rối loạn, nhưng chỉ có sẵn dưới dạng dịch vụ chẩn đoán thông qua các phòng thí nghiệm chuyên biệt.
Có thể chẩn đoán trước sinh ở những thai kỳ có nguy cơ cao bị phù thai do Hb Bart bằng siêu âm Doppler, một thủ thuật không xâm lấn trong đó sóng âm thanh phản xạ được sử dụng để tạo ra hình ảnh của thai nhi đang phát triển cho phép các bác sĩ xem máu chảy qua các mạch máu như thế nào. Cụ thể, xét nghiệm này được sử dụng để đo tốc độ lưu lượng máu qua các động mạch não của thai nhi, tỷ lệ này tương quan chặt chẽ với tình trạng thiếu máu ở thai nhi. Ở những thai kỳ có nguy cơ cao, thai nhi bị phù nước Hb Bart có thể được chẩn đoán sớm nhất là vào tuần thứ 13 đến 14 của thai kỳ.
Việc xét nghiệm các thành viên trực hệ trong gia đình như những đứa trẻ khác hoặc anh chị em của cha mẹ bị ảnh hưởng được khuyến nghị bởi vì, ngay cả khi không có triệu chứng, những cá nhân này có thể là người mang mầm bệnh alpha thalassemia thể thầm lặng hoặc bệnh alpha thalassemia thể nhẹ.
Tài liệu tham khảo
- De Alarcon PA, Werner EJ, Christensen RD. Eds. Alpha Thalassemia. In: Neonatal Hematology: Pathogenesis, Diagnosis, and Management of Hematologic Problems. 2nd ed. Cambridge University Press, New York, NY; 2013:105-107.
- Piel FB, Weatherall DJ. The alpha-thalassemias. N Engl J Med. 2014;371:1908-1916. http://www.ncbi.nlm.nih.gov/pubmed/25390741
- Vichinsky EP. Clinical manifestations of α-thalassemia. Cold Spring Harb Perspect Med. 2013;3:a011742. http://www.ncbi.nlm.nih.gov/pubmed/23543077
- Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med. 2011;13:83-88. http://www.ncbi.nlm.nih.gov/pubmed/21381239
- Harteveld CL, Higgs DR. α-thalassemia. Orphanet J Rare Dis. 2010;5:13. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2887799/
(*) Theo NORD