Trong những năm gần đây, công nghệ giải trình tự gen đã đạt được những bước tiến vượt bậc. NGS (Next-Generation Sequencing), hay còn gọi là công nghệ giải trình tự gen thế hệ mới, đã ra đời và nhanh chóng trở thành công cụ đắc lực cho các nhà khoa học, các chuyên gia mở ra những cánh cửa mới cho nghiên cứu di truyền.
Vậy công nghệ giải trình tự gen thế hệ mới (NGS) là gì? Tham khảo bài viết NOVAGEN chia sẻ sau để tìm hiểu chi tiết!
 là gì?")
Nội dung:
1. Giới thiệu về phương pháp và kỹ thuật NGS
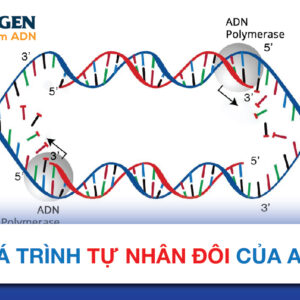
Giải trình tự thế hệ tiếp mới (NGS) là công nghệ xác định trình tự ADN hoặc ARN để nghiên cứu biến thể di truyền liên quan đến bệnh tật hoặc các hiện tượng sinh học khác. Được giới thiệu để sử dụng thương mại vào năm 2005, phương pháp này ban đầu được gọi là “giải trình tự song song hàng loạt”, vì nó cho phép giải trình tự nhiều sợi ADN cùng lúc, thay vì từng sợi một như giải trình tự Sanger truyền thống bằng phương pháp điện di mao quản (CE).
Mỗi công nghệ đều mang lại những tiện ích khác nhau trong môi trường phân tích di truyền ngày nay. Giải trình tự Sanger là phương pháp tốt nhất để phân tích số lượng nhỏ các mục tiêu gen, mẫu và có thể hoàn thành trong một ngày. Nó cũng được coi là công nghệ giải trình tự tiêu chuẩn vàng nên kết quả NGS thường được xác minh bằng cách sử dụng giải trình tự Sanger.
NGS cho phép thẩm vấn hàng trăm đến hàng nghìn gen cùng một lúc trong nhiều mẫu, cũng như khám phá và phân tích các loại đặc điểm bộ gen khác nhau trong một lần chạy giải trình tự, từ các biến thể nucleotide đơn (SNV) đến số lượng bản sao và các biến thể cấu trúc, thậm chí cả hợp nhất ARN. NGS cho phép thông lượng lý tưởng cho mỗi lần chạy nên các nghiên cứu có thể được thực hiện nhanh chóng, tiết kiệm chi phí hơn. Các lợi thế bổ sung của NGS bao gồm yêu cầu đầu vào mẫu thấp hơn, độ chính xác cao hơn và khả năng phát hiện các biến thể ở tần số alen thấp hơn so với giải trình tự Sanger.
Tốc độ, thông lượng và độ chính xác của NGS đã cách mạng hóa phân tích di truyền, cho phép ứng dụng vào trong các nghiên cứu bộ gen và lâm sàng, sức khỏe sinh sản, khoa học môi trường, nông nghiệp, pháp y.
2. Các bước trong quy trình công nghệ giải trình tự gen thế hệ mới

2.1. Chuẩn bị thư viện NGS – Công nghệ giải trình tự gen thế hệ mới
Một “thư viện” giải trình tự phải được tạo ra từ mẫu. Mẫu ADN (hoặc cADN) được xử lý thành các đoạn mạch kép tương đối ngắn (100–800 bp). Tùy thuộc vào ứng dụng cụ thể, quá trình phân mảnh ADN có thể được thực hiện theo nhiều cách khác nhau, bao gồm cắt vật lý, tiêu hóa bằng enzyme và khuếch đại dựa trên PCR các vùng di truyền cụ thể. Các đoạn ADN thu được sau đó được nối với các trình tự bộ điều hợp công nghệ cụ thể, tạo thành một thư viện đoạn. Các bộ điều hợp này cũng có thể có một “mã vạch” phân tử duy nhất, do đó, mỗi mẫu có thể được gắn thẻ bằng một trình tự ADN duy nhất. Điều này cho phép trộn nhiều mẫu lại với nhau và giải trình tự cùng một lúc. Ví dụ, có thể sử dụng mã vạch 1-20 để dán nhãn riêng cho 20 mẫu và sau đó phân tích chúng trong một lần giải trình tự duy nhất. Phương pháp này, được gọi là “gộp” hoặc “ghép kênh”, giúp tiết kiệm thời gian, tiết kiệm chi phí trong các thí nghiệm giải trình tự và kiểm soát sự thay đổi của quy trình làm việc, vì các mẫu gộp được xử lý cùng nhau.
Ngoài các thư viện đoạn, còn có hai phương pháp chuyên biệt khác để chuẩn bị thư viện: thư viện đầu ghép và thư viện cặp đôi. Thư viện đầu ghép cho phép người dùng giải trình tự đoạn ADN từ cả hai đầu, thay vì giải trình tự thông thường chỉ diễn ra theo một hướng duy nhất. Thư viện đầu ghép được tạo giống như các thư viện đoạn thông thường, nhưng chúng có thẻ bộ điều hợp ở cả hai đầu của đoạn chèn ADN cho phép giải trình tự theo hai hướng. Phương pháp này giúp lập bản đồ các đoạn đọc dễ dàng hơn và có thể được sử dụng để cải thiện khả năng phát hiện sắp xếp lại bộ gen, các thành phần trình tự lặp lại và các hợp nhất gen RNA hoặc các biến thể ghép nối. Tuy nhiên, những cải tiến trong các phương pháp chuẩn bị thư viện hiện đại và các công cụ phân tích đã giúp phát hiện các tính năng này bằng giải trình tự một hướng.
Thư viện cặp đôi có cách tạo phức tạp hơn so với thư viện đoạn hoặc cặp đôi và có liên quan đến các đoạn chèn ADN có kích thước lớn hơn nhiều (trên 2Kb và lên đến 30Kb). Giải trình tự thư viện cặp đôi tạo ra hai đoạn đọc cách xa nhau và theo hướng ngược nhau. Sử dụng thông tin vật lý liên quan giữa hai đoạn đọc giải trình tự, giải trình tự cặp đôi hữu ích cho lắp ráp de novo, phát hiện biến thể cấu trúc lớn và xác định các sắp xếp lại bộ gen phức tạp.
2.2. Khuếch đại dòng vô tính cho NGS
Trước khi giải trình tự, thư viện ADN phải được gắn vào một bề mặt rắn và được khuếch đại theo kiểu nhân bản để tăng tín hiệu có thể phát hiện được từ mỗi mục tiêu trong quá trình giải trình tự. Trong quá trình này, mỗi phân tử ADN riêng biệt trong thư viện được gắn vào bề mặt của một hạt hoặc một tế bào dòng chảy và được khuếch đại PCR để tạo ra một tập hợp các bản sao giống hệt nhau. Trong trường hợp của công nghệ Ion Torrent, một quy trình được gọi là “tạo mẫu” được sử dụng để thêm các phân tử thư viện vào các hạt.
2.3. Thực hiện giải trình tự thư viện NGS
Tất cả ADN trong thư viện được giải trình tự cùng lúc bằng một thiết bị giải trình tự. Mặc dù mỗi công nghệ NGS là duy nhất, tất cả đều sử dụng một phiên bản của phương pháp “giải trình tự bằng tổng hợp”, đọc từng bazơ khi chúng phát triển dọc theo một sợi đã trùng hợp. Đây là một chu trình với các bước chung: tổng hợp bazơ ADN trên ADN sợi đơn, tiếp theo là phát hiện bazơ đã kết hợp, và sau đó loại bỏ các chất phản ứng để khởi động lại chu trình.
Hầu hết các thiết bị giải trình tự sử dụng phương pháp phát hiện quang học để xác định sự kết hợp nucleotide trong quá trình tổng hợp ADN. Các thiết bị Ion Torrent sử dụng phương pháp phát hiện điện để cảm nhận sự giải phóng các ion hydro, điều này xảy ra tự nhiên khi các nucleotide được kết hợp trong quá trình tổng hợp ADN.
2.4. Phân tích dữ liệu giải trình tự thế hệ tiếp theo
Mỗi thí nghiệm NGS tạo ra một lượng lớn dữ liệu phức tạp bao gồm các đoạn đọc ADN ngắn. Mặc dù mỗi nền tảng công nghệ có thuật toán và công cụ phân tích dữ liệu riêng, nhưng chúng chia sẻ một ‘đường ống’ phân tích tương tự và sử dụng các số liệu chung để đánh giá chất lượng của các tập dữ liệu NGS.

Phân tích có thể được chia thành ba bước: phân tích sơ cấp, thứ cấp và bậc ba. Phân tích sơ cấp là quá trình xử lý các tín hiệu thô từ các máy dò công cụ thành dữ liệu số hóa hoặc các lệnh gọi cơ sở. Các dữ liệu thô này được thu thập trong mỗi chu kỳ giải trình tự. Đầu ra của phân tích sơ cấp là các tệp chứa các lệnh gọi cơ sở được lắp ráp thành các lần đọc giải trình tự (tệp FASTQ) và điểm chất lượng liên quan của chúng (điểm chất lượng Phred). Phân tích thứ cấp bao gồm lọc và cắt xén đọc dựa trên chất lượng, sau đó là căn chỉnh các lần đọc với bộ gen tham chiếu hoặc lắp ráp các lần đọc cho bộ gen mới và cuối cùng là gọi biến thể. Đầu ra chính là tệp BAM chứa các lần đọc đã căn chỉnh. Phân tích bậc ba là bước đầy thách thức nhất vì nó liên quan đến việc diễn giải kết quả và trích xuất thông tin có ý nghĩa từ dữ liệu.
Tham khảo: Thermo Fisher Scientific