Xét Nghiệm Gen Di Truyền HBA1 Và HBA2 Chẩn Đoán Bệnh Alpha Thalassemia. Các biến thể gây bệnh (thường là mất đoạn) trong các gen này gây ra bệnh Alpha thalassemia.
Bài viết này thảo luận về ý nghĩa của kết quả xét nghiệm di truyền đối với gen alpha globin (HBA1 và HBA2). Các biến thể gây bệnh (thường là mất đoạn) trong các gen này gây ra bệnh Alpha thalassemia.
Xét nghiệm gen alpha globin không thể xác định bệnh Beta thalassemia hoặc các tình trạng khác ảnh hưởng đến beta globin chẳng hạn như bệnh hồng cầu hình liềm.
Nội dung:
- 1 Đặc điểm di truyền bệnh Alpha thalassemia
- 2 Khi nào cần làm xét nghiệm gen di truyền HBA1 và HBA2
- 3 Kết quả xét nghiệm gen di truyền HBA1 và HBA2
- 4 Xét nghiệm chẩn đoán bệnh Alpha Thalassemia
- 5 Các phương pháp điều trị và hỗ trợ điều trị bệnh Alpha thalassemia
- 6 Tư vấn sinh sản đối với bệnh Alpha thalassmia
Đặc điểm di truyền bệnh Alpha thalassemia
Huyết sắc tố (Hb) là một tetramer của 2 chuỗi alpha globin và 2 chuỗi beta globin.
Bệnh Alpha thalassemia gây ra bởi các biến thể gây bệnh trong gen alpha globin, dẫn đến giảm sản xuất alpha globin.
Có 4 gen alpha globin chức năng (hai gen riêng biệt, liền kề trong cụm gen alpha globin được thừa hưởng từ mỗi cha mẹ) (hình 1).
- Bất kỳ số lượng gen alpha globin nào từ 1 đến 4 đều có thể bị ảnh hưởng.
- Càng nhiều chuỗi alpha bị ảnh hưởng, hội chứng Alpha thalassemia càng nặng.
Mặc dù beta thalassemia có thể quen thuộc hơn, nhưng các biến thể alpha thalassemia phổ biến hơn trên toàn thế giới.
- Biến thể alpha 0 (biến thể xóa) hủy bỏ quá trình sản xuất alpha globin;
- Biến thể alpha + (biến thể không xóa, đôi khi được chỉ định là ND) tạo ra một số alpha globin nhưng ít hơn bình thường.
Mặc dù một số biến thể alpha + là nhẹ, nhưng những biến thể khác có thể nghiêm trọng hoặc tệ hơn biến thể xóa.
* Các biến thể alpha 0 – Thường xóa toàn bộ cụm gen alpha globin.
* Các biến thể alpha + – Thay đổi một hoặc một vài nucleotide. Hemoglobin Constant Spring (Hb CS) là một biến thể không xóa phổ biến, gây ra bởi đột biến của bộ ba mã hóa cuối cùng trong gen alpha2 globin.
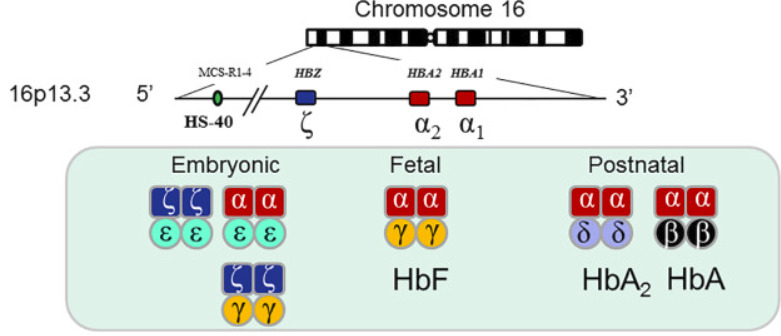
Hình 2: Vùng gen mã hóa alpha và beta globin
Quá trình sản xuất alpha globin bắt đầu từ trong tử cung (hình 2).
Ở bào thai, alpha globin kết hợp với gamma globin để tạo ra huyết sắc tố bào thai (Hb F); sản xuất beta globin bắt đầu sau khi sinh.
Do đó, các biến thể alpha globin biểu hiện trong tử cung, trong khi các biến thể beta globin biểu hiện sau sáu tháng tuổi.
Tỷ lệ của chuỗi alpha và beta phải được kết hợp chặt chẽ.
Nếu một loại chuỗi bị dư thừa, nó sẽ tạo thành chất kết tủa trong hồng cầu hoặc tiền chất hồng cầu trong tủy xương.
Những Hbs kết tủa này bao gồm Hb Barts (bộ tứ của gamma globin) và Hb H (bộ tứ của beta globin).
Những Hb bất thường này làm tăng mức Hb nhưng vô dụng về mặt chức năng vì chúng không vận chuyển oxy. Các chất kết tủa thúc đẩy tan máu.
Khi nào cần làm xét nghiệm gen di truyền HBA1 và HBA2
Xét nghiệm gen alpha globin được sử dụng khi:
- Chẩn đoán bệnh Alpha thalassemia.
- Xét nghiệm tiền hôn nhân
- Xét nghiệm sàng lọc trước sinh không xâm lấn
- Thực hiện hỗ trợ sinh sản IVF
Kết quả xét nghiệm gen di truyền HBA1 và HBA2
Các đặc điểm lâm sàng bao gồm từ trạng thái mang mầm bệnh không có triệu chứng đến tử vong trong tử cung, tùy thuộc vào số lượng gen alpha globin bị ảnh hưởng và liệu các biến thể là xóa hay không xóa.
* Một gen alpha thalassemia – Những cá nhân có một biến thể/xóa gen alpha duy nhất (alpha thalassemia cực tiểu hoặc người mang mầm bệnh thầm lặng) không có triệu chứng mà không có bất kỳ thay đổi nào trong phòng xét nghiệm.
* Hai gen alpha thalassemia – Những người có hai biến thể/xóa gen alpha (alpha thalassemia thể nhẹ hoặc đặc điểm alpha thalassemia) không có triệu chứng. Họ có thể bị thiếu máu vi mô nhẹ hoặc thiếu máu vi mô mà không bị thiếu máu. Huyết sắc tố (Hb) giảm khoảng 1 g/dL; nó có thể giảm hơn nữa trong thời kỳ mang thai. Hai kiểu gen có thể xảy ra là dị hợp tử đối với kiểu gen alpha 0 (aa/–) hoặc dị hợp tử đối với kiểu gen alpha + (a-/a-). Những điều này thường không ảnh hưởng đến tình trạng lâm sàng của cá nhân, nhưng chúng có thể quan trọng đối với tư vấn sinh sản.
* Ba gen alpha thalassemia – Bệnh Hb H đề cập đến ba lần xóa gen alpha (a-/–) hoặc các biến thể như (a-/-a CS ). Hb H là một tetrame của beta globin.
- Những người mắc bệnh mất đoạn Hb H bị thiếu máu từ nhẹ đến trung bình và đôi khi cần truyền máu. Lách to và sỏi sắc tố là phổ biến. Thiếu máu có thể trở nên trầm trọng hơn do các bệnh do virus thông thường và tiếp xúc với thuốc oxy hóa, với yêu cầu truyền máu thoáng qua đột ngột.
- Những người mắc bệnh Hb H không mất đoạn như (a-/-a CS ) bị thiếu máu trầm trọng hơn nhưng ít tế bào vi mô hơn. Họ có thể yêu cầu truyền máu trong tử cung hoặc khi sinh; một số có thể yêu cầu truyền máu sau khi sinh. Một số trẻ sơ sinh bị Hb CS phát triển lách to rõ rệt và các triệu chứng nghiêm trọng cần truyền máu mãn tính trong thời gian dài; có tới 20 phần trăm bệnh nhân mắc Hb CS cần truyền máu và cắt lách.
Những người có ba biến thể gen alpha thalassemia có thể tạo hồng cầu không hiệu quả và tăng hấp thu sắt, đồng thời có thể bị quá tải sắt do truyền máu hoặc không do truyền máu.
* Bốn gen alpha thalassemia – Biến thể/xóa cả bốn gen alpha globin (–/–) gây ra bệnh alpha thalassemia thể nặng, dạng nghiêm trọng nhất của bệnh alpha thalassemia. Hầu hết Hb tuần hoàn là Hb Barts, không vận chuyển oxy. Những người này phát triển thai nhi phù nước và thường chết trong tử cung trừ khi họ được truyền máu trong tử cung. Sau khi sinh, chúng cần được truyền máu mãn tính bằng liệu pháp thải sắt. Ghép tế bào gốc tạo máu allogeneic (Hematopoietic Stem Cell Transplant – HSCT) có khả năng chữa bệnh.
Xét nghiệm chẩn đoán bệnh Alpha Thalassemia
Tất cả những người nghi ngờ mắc bệnh alpha thalassemia nên được xét nghiệm công thức máu toàn bộ (Complete Blood Count – CBC) và xem xét các chỉ số hồng cầu (RBC).
Xét nghiệm tán huyết là thích hợp nếu có thiếu máu hoặc tăng hồng cầu lưới.
* Xét nghiệm công thức máu toàn phần – Thể tích hồng cầu trung bình (MCV) <80 fL và/hoặc huyết sắc tố trung bình trong hồng cầu (MCH) <27 phù hợp với bệnh alpha thalassemia, bất kể có thiếu máu hay không, nhưng chúng không được chẩn đoán, vì chúng cũng có thể xảy ra khi thiếu sắt và các rối loạn khác.
* Xét nghiệm sắt – Những người được đánh giá bệnh thalassemia nên được xét nghiệm sắt để loại trừ hoặc chẩn đoán tình trạng thiếu sắt và để đánh giá tình trạng quá tải sắt.
- Thiếu sắt gây thiếu máu hồng cầu nhỏ và có thể cản trở quá trình phân tích huyết sắc tố dựa trên protein.
- Tình trạng ứ sắt thường gặp ở bệnh nhân thalassemia.
* Phân tích huyết sắc tố dựa trên protein hoặc ADN – Việc xác nhận chẩn đoán có thể được thực hiện bằng xét nghiệm phân tử hoặc dựa trên protein.
- Xét nghiệm dựa trên protein chẳng hạn như sắc ký lỏng hiệu năng cao (HPLC) hoặc điện di (dựa trên gel hoặc mao quản) có thể xác nhận bệnh alpha thalassemia nếu có Hb H (hoặc Hb Barts). HPLC hoặc điện di mao quản được ưa thích hơn; chúng chính xác và định lượng hơn. Các phương pháp dựa trên protein có thể phát hiện Hb A2 và Hb F, có thể hỗ trợ nhưng ít đặc hiệu hơn. Hb A2 >4% là chẩn đoán beta thalassemia; Hb A2 thấp gợi ý bệnh alpha thalassemia hoặc thiếu sắt.
- Xét nghiệm sinh học phân tử (ADN) cung cấp kiểu gen cụ thể. Đối với bệnh Alpha thalassemia cực tiểu (một gen alpha globin bị ảnh hưởng) cần tới xét nghiệm phân tử kiểu này.
Phân tích Hb có thể được bỏ qua trong một số trường hợp, chẳng hạn như những người có CBC bình thường không có ý định sinh con.
Các phương pháp điều trị và hỗ trợ điều trị bệnh Alpha thalassemia
* Bổ sung Axit folic – Tất cả những người bị tan máu mãn tính nên dùng axit folic (liều điển hình, 1 mg mỗi ngày; liều cao hơn trước khi thụ thai và trong khi mang thai). Điều này có thể được bỏ qua một cách hợp lý ở những người có chế độ ăn giàu folate và không cố gắng thụ thai.
* Truyền máu – Có thể cần truyền máu cho bệnh thiếu máu nặng, khủng hoảng bất sản thoáng qua hoặc các yếu tố gây căng thẳng khác cho tủy xương.
* Cắt túi mật/cắt lách – Cắt túi mật có thể cần thiết trong bệnh Hb H do sỏi mật sắc tố. Bệnh Hb H với một biến thể không xóa (non-deletional variant) thường phải cắt lách để cải thiện nồng độ Hb.
* Thải sắt – Khi bệnh nhân Hb H già đi, họ bị quá tải sắt ngay cả khi không truyền máu; họ cần được yêu cầu theo dõi và tăng cường khả năng thải sắt.
* Chất kích hoạt PK – Mitapivat là một chất kích hoạt phân tử nhỏ của pyruvate kinase (PK) làm tăng huyết sắc tố trong một nghiên cứu nhỏ; các thử nghiệm tiếp theo đang diễn ra.
* Alpha thalassemia thể nặng – Alpha thalassemia thể nặng cần điều trị tích cực, truyền máu trong tử cung, sau đó là truyền máu suốt đời và thải sắt. Ghép tế bào gốc tạo máu (HSCT) có thể chữa khỏi bệnh; HSCT trong tử cung thai nhi đang được điều tra. Khả năng HSCT sau khi sinh nên được thảo luận.
Tư vấn sinh sản đối với bệnh Alpha thalassmia
Những người mắc bệnh alpha thalassemia nên được tư vấn sinh sản phù hợp với lứa tuổi. Những người có kế hoạch thụ thai nên được bác sĩ Sản khoa tư vấn cụ thể.
Các biến thể alpha thalassemia phổ biến ở những người có tổ tiên từ miền Nam Trung Quốc, Malaysia, Thái Lan và Châu Phi. Tuy nhiên, do di cư, các biến thể alpha thalassemia có thể xuất hiện ở những cá nhân thuộc bất kỳ tổ tiên nào.
- Dạng alpha thalassemia-1 (xóa hai gen alpha trong cis; aa/–) phổ biến hơn ở những người có tổ tiên từ Nam Trung Quốc, Malaysia và Thái Lan; những cá nhân này có nguy cơ cao hơn khi mang thai bị phù thai nhi (Hb Barts).
- Dạng alpha thalassemia-2 (xóa hai gen alpha trong trans; a-/a-) thường gặp ở những người có nguồn gốc châu Phi thường mang đặc điểm. Các biến thể gây bệnh nằm trên các nhiễm sắc thể đối diện và bất kể các biến thể của người cha là gì, thai nhi có bốn biến thể gây bệnh (Hb Barts) là không thể. Hb H vẫn có thể tùy thuộc vào kiểu gen của đối tác.
Những thai kỳ có biến thể nghiêm trọng có nguy cơ thai nhi bị ảnh hưởng cao hơn và những người mang kiểu gen alpha 0 (aa/–) có nguy cơ sinh con mắc bệnh alpha thalassemia thể nặng. Họ có thể lựa chọn:
- Xét nghiệm di truyền tiền làm tổ (PGT) trên phôi được tạo ra bằng phương pháp thụ tinh trong ống nghiệm (IVF).
- Sử dụng tế bào trứng của người hiến tặng hoặc tinh trùng của người hiến tặng.
- Nhận con nuôi.
- Xét nghiệm tiền sản có theo dõi chuyên sâu và điều trị trong tử cung nếu cần.
(*) Theo UpToDate
*** Bài viết chỉ có giá trị tham khảo. Không thay thế chẩn đoán và điều trị của bác sĩ ***