Sinh Lý Học Bệnh Thalassemia Gây Tan Máu Bẩm Sinh
Thalassemia đề cập đến một nhóm bệnh di truyền gen lặn trên nhiễm sắc thể thường, được đặc trưng bởi sự giảm hoặc không sản xuất một hoặc nhiều chuỗi globin. Beta thalassemia là do suy giảm sản xuất chuỗi beta globin; alpha thalassemia là do suy giảm sản xuất chuỗi alpha globin.
Alpha và beta thalassemia là các bệnh huyết sắc tố di truyền, trong đó việc sản xuất một loại chuỗi globin bị suy giảm (chuỗi alpha globin trong bệnh alpha thalassemia; chuỗi beta globin trong bệnh thalassemia beta) gây ra sự mất cân bằng về tỷ lệ giữa chuỗi alpha và beta (hoặc giống beta).
Sự tổng hợp cân bằng là rất quan trọng vì các phân tử tetramer huyết sắc tố A (Hb A) và huyết sắc tố F (Hb F) nguyên vẹn có khả năng hòa tan cao trong tế bào chất của tế bào hồng cầu (Red Blood Cell – RBC), nhưng các chuỗi alpha và beta không liên kết thì không. Chúng kết tủa, gây tổn thương tế bào đáng kể.
Những chuỗi globin không ghép cặp này gây ra các vấn đề với sự trưởng thành của hồng cầu và dẫn đến quá trình tạo hồng cầu không hiệu quả, thiếu máu tán huyết, quá tải sắt và các biến chứng tiếp theo.
Beta Thalassemia
Beta thalassemia gây ra bởi các biến thể (đột biến) trong locus huyết sắc tố beta (gen beta globin, kí hiệu là HBB) dẫn đến suy giảm sản xuất chuỗi beta globin. Điều này dẫn đến sự dư thừa chuỗi alpha hoặc chuỗi loại alpha.
Các biến thể beta 0 là những biến thể hủy bỏ quá trình sản xuất beta globin và các biến thể beta + có thể tạo ra một số beta globin nhưng ít hơn nhiều so với lượng bình thường. Kiểu hình lâm sàng phụ thuộc vào sự kết hợp của các biến thể beta globin mà một cá nhân mang.
Trước đây, những người mắc bệnh beta thalassemia được phân loại (theo thứ tự mức độ nghiêm trọng giảm dần) thành bệnh beta thalassemia thể nặng, trung bình và nhẹ.
Việc phân loại sau đó đã chuyển sang sử dụng thuật ngữ phụ thuộc truyền máu (transfusion dependent) và không phụ thuộc truyền máu (non-transfusion dependent).
Thuật ngữ mới đã được thông qua vì dựa trên tình trạng lâm sàng của bệnh nhân liên quan đến một đặc điểm tiên lượng chính (phụ thuộc truyền máu suốt đời, hoặc không) và phân biệt rõ ràng hai nhóm bệnh nhân này.
Ngược lại, dạng bệnh thalassemia thể trung gian được cho là quá rộng và mơ hồ, từ triệu chứng tối thiểu đến dấu hiệu lâm sàng trùng lặp với bệnh thalassemia thể beta nặng phụ thuộc vào truyền máu.
Phụ thuộc vào truyền máu – Những người phụ thuộc vào truyền máu cần được truyền máu thường xuyên để giảm thiểu tình trạng thiếu máu trầm trọng và tạo máu ngoài tủy. Những cá nhân này trước đây được gọi là mắc bệnh beta thalassemia thể nặng. Những người cần truyền máu định kỳ vì bệnh thiếu máu không được coi là phụ thuộc vào truyền máu, nhưng họ có thể trở nên như vậy theo thời gian.
Không phụ thuộc vào truyền máu – Những người không phụ thuộc vào truyền máu không cần truyền máu thường xuyên. Họ có thể không bao giờ được truyền máu, hoặc họ có thể yêu cầu truyền máu định kỳ do thiếu máu (ví dụ: trong khi mang thai hoặc nhiễm trùng cấp tính). Những người này được mô tả là mắc bệnh thalassemia thể trung gian nếu thiếu máu và/hoặc có dấu hiệu thiếu máu tán huyết hoặc quá trình tạo hồng cầu không hiệu quả, và là mắc bệnh thalassemia thể nhẹ nếu ít có triệu chứng. Tuy nhiên, thuật ngữ thalassemia thể nhẹ cũng được sử dụng để mô tả trạng thái dị hợp tử hầu như không có triệu chứng. Sự nhầm lẫn này là một lý do bổ sung để chuyển sang thuật ngữ mới.
Mỗi cá nhân có hai gen beta globin (một gen từ bố và mẹ). Các biến thể gen beta thalassemia được phân loại theo mức độ giảm sản xuất beta globin:
* Beta 0 thalassemia: đề cập đến các đột biến làm mất hoàn toàn quá trình sản xuất beta globin. Những bệnh nhân đồng hợp tử hoặc dị hợp tử về đột biến beta 0 thalassemia có nhiều khả năng mắc bệnh beta thalassemia phụ thuộc vào truyền máu (trước đây gọi là beta thalassemia thể nặng); chúng không thể tạo ra bất kỳ chuỗi beta globin nào và do đó không thể tạo ra huyết sắc tố trưởng thành bình thường (hemoglobin A [Hb A]; alpha2/beta2). Đây là những bệnh nhân được Cooley mô tả ban đầu. Họ thường bị thiếu máu trầm trọng, phụ thuộc vào truyền máu và các biểu hiện khác của bệnh thalassemia nặng. Những người dị hợp tử về beta 0 thalassemia có nhiều khả năng có huyết sắc tố thấp hơn, và một số thậm chí có thể biểu hiện dấu hiệu tan máu nhẹ và tạo hồng cầu không hiệu quả.
Có những trường hợp ngoại lệ đối với xu hướng chung này và bệnh nhân thalassemia beta 0 có thể không phụ thuộc vào truyền máu. Ví dụ, một số đột biến hủy bỏ quá trình tổng hợp beta globin, chẳng hạn như delta beta thalassemia (còn gọi là hội chứng Hb Lepore) có liên quan đến việc sản xuất bù đáng kể chuỗi gamma, và do đó, huyết sắc tố thai nhi (Hb F) trong cuộc sống sau khi sinh. Sự đồng thời của đặc điểm alpha thalassemia cũng có thể làm giảm sự mất cân bằng chuỗi, do đó làm giảm mức độ nghiêm trọng.
* Beta + thalassemia: đề cập đến các đột biến làm giảm (nhưng không có) sản xuất beta globin. Những bệnh nhân đồng hợp tử về đột biến beta + thalassemia có thể tạo ra một số Hb A và có xu hướng ít bị ảnh hưởng nghiêm trọng hơn so với những người có đột biến beta 0 thalassemia.
– Những người có đột biến beta + thalassemia kết hợp với đột biến beta 0 thalassemia thường mắc bệnh beta thalassemia phụ thuộc vào truyền máu.
– Những người có hai đột biến beta + thalassemia thường mắc bệnh beta thalassemia không phụ thuộc vào truyền máu, mặc dù một số người có thể phụ thuộc vào truyền máu hoặc có thể trở nên phụ thuộc vào truyền máu sau này trong đời.
– Những người dị hợp tử về bệnh beta + thalassemia thừa hưởng alen beta thalassemia từ cha hoặc mẹ và alen beta globin bình thường từ cha hoặc mẹ còn lại. Những cá nhân này bị bệnh tiểu cầu nghiêm trọng nhưng thiếu máu nhẹ hoặc tối thiểu; chúng phần lớn không có triệu chứng và thường được xác định tình cờ (bằng xét nghiệm gia đình hoặc xét nghiệm công thức máu toàn bộ [CBC] được thực hiện vì những lý do khác). Thiếu máu có thể trở nên nghiêm trọng hơn trong thai kỳ.
Alpha thalassemia
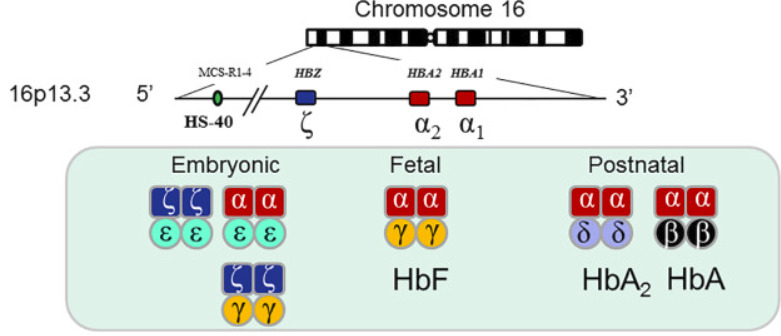
Alpha thalassemia gây ra bởi đột biến ở locus alpha haemoglobin (gen alpha globin) dẫn đến suy giảm khả năng sản xuất chuỗi alpha globin (Hình 1).
Điều này dẫn đến sự dư thừa tương đối của chuỗi beta và chuỗi giống beta. Phụ thuộc phần lớn vào kiểu gen alpha globin, những người mắc bệnh alpha thalassemia có triệu chứng có thể không phụ thuộc vào việc truyền máu (phổ biến) hoặc phụ thuộc vào truyền máu (hiếm gặp).
Các biến thể alpha 0 là những biến thể hủy bỏ quá trình sản xuất alpha globin và các biến thể alpha + có thể tạo ra một số alpha globin nhưng ít hơn lượng bình thường.
- Biến thể alpha 0 là biến thể trong đó toàn bộ cụm gen alpha globin bị xóa (biến thể xóa);
- Biến thể alpha + có thể liên quan đến đột biến điểm (biến thể không xóa). Biến thể alpha globin không xóa phổ biến nhất là Hemoglobin Constant Spring.
Ngược lại với bệnh beta thalassemias, trong đó các biến thể beta + có xu hướng ít nghiêm trọng hơn, một số alen alpha + như Hb Constant Spring thực sự có thể làm trầm trọng thêm mức độ nghiêm trọng. Điều này là do alen alpha + phổ biến mã hóa chuỗi alpha, khi kết hợp với chuỗi beta, tạo ra các huyết sắc tố không ổn định cao kết tủa, làm tăng thêm gánh nặng của các thể vùi trong hồng cầu đang phát triển và lưu thông.
Cụm gen alpha globin có hai bản sao của gen alpha globin, alpha2 và alpha1. Do đó, mỗi cá nhân có bốn gen alpha globin (hai gen từ bố và mẹ); ký hiệu “aa/aa” được sử dụng để mô tả bốn gen alpha globin hoạt động bình thường. Kiểu hình lâm sàng phụ thuộc vào sự kết hợp của các biến thể alpha globin mà một cá nhân mang. Có thể có nhiều biến thể kết hợp, làm tăng độ phức tạp của việc phân loại bệnh alpha thalassemia.
* Phù thai nhi với Hb Barts – Hội chứng phù thai nhi (Hydrops fetalis) là dạng nghiêm trọng nhất của bệnh alpha thalassemia, do xóa hoặc bất hoạt cả bốn gen alpha globin (–/–). Hội chứng này trước đây được coi là không tương thích với sinh sống, do thiếu chuỗi alpha thường xuất hiện sau tuần thứ năm hoặc thứ sáu của thai nhi. Tuy nhiên, các ca sinh sống của trẻ sơ sinh khỏe mạnh đã được báo cáo với chẩn đoán trước sinh và truyền máu trong tử cung, sau đó trong một số trường hợp ghép tủy xương dị loài.
Hemoglobin bào thai bình thường (Hb F) là một tetramer của chuỗi alpha và chuỗi gamma. Trong trường hợp không có chuỗi alpha chức năng, chuỗi gamma homotetramer hóa để tạo thành Hemoglobin Barts (Hb Barts). Hb Barts ít có xu hướng kết tủa và tạo thành thể vùi. Tuy nhiên, Hb Barts (cũng như homotetramers của beta globin; hemoglobin H [Hb H]) về mặt chức năng là vô dụng đối với việc cung cấp oxy do ái lực cao của chúng đối với oxy (gấp 10 lần so với Hb A); chúng dễ bị tổn thương do oxy hóa. Chúng không có hiệu ứng Bohr, và chúng thiếu đường cong phân ly hemoglobin-oxy hình sigmoidal do thiếu tương tác heme-heme. Thay vào đó, đường cong phân ly oxyhemoglobin của chúng giống với đường cong của myoglobin.
Chuỗi Zeta là một chuỗi giống như phôi alpha. Nếu khiếm khuyết phân tử cho phép thai nhi tổng hợp chuỗi zeta, thì huyết sắc tố Portland (Hb Portland; zeta2 gamma2) có thể được tạo ra, cho phép sự sống sót trong tử cung cho đến tam cá nguyệt thứ ba. Những thai nhi này bị thiếu máu nghiêm trọng về mặt chức năng, với suy tim cung lượng cao, bệnh thoát vị đĩa đệm và rò rỉ mao mạch (“thủy dịch”), cùng với nhau thai to, tăng huyết áp và đa ối ở mẹ, mặc dù thực tế là nồng độ huyết sắc tố đo được có thể cao tới 10 g /dL do sự hiện diện của Hb Barts và Hb Portland (xem phần “Thai nhi phù nước không do miễn dịch”). Các tế bào hồng cầu (RBCs) là hypochromic và microcytic, và quá trình tạo hồng cầu được mở rộng rất nhiều để bù đắp cho tình trạng thiếu oxy mô sâu dẫn đến, với sự tạo hồng cầu ngoài tủy bù trừ ở gan và lá lách. Một thành phần nhỏ của Hb H cũng có thể xuất hiện trong giai đoạn cuối của thai kỳ, khi bào thai bắt đầu tổng hợp một lượng nhỏ chuỗi beta.
* Bệnh Hb H – Bệnh Hb H (Hemoglobin H disease) đề cập đến việc xóa hoặc bất hoạt ba trong số bốn gen alpha globin (a-/–). Sự tổng hợp chuỗi globin không cân bằng có ý nghĩa lâm sàng, với tỷ lệ tổng hợp alpha/beta từ 0,3 đến 0,6 (bình thường, 1,0 ± 0,05), gây ra sự tích tụ chuỗi gamma không ghép cặp trong quá trình phát triển của bào thai và giai đoạn đầu của trẻ sơ sinh và sự tích tụ chuỗi beta không ghép cặp ở tuổi trưởng thành.
Kết quả là homotetramer gamma globin ở bào thai và trẻ sơ sinh tạo thành Hb Barts, và kết quả là homotetramer beta globin ở trẻ em và người lớn ở dạng Hb H. Như đã lưu ý ở trên, Hb Barts và Hb H không có chức năng vận chuyển oxy. Tuy nhiên, sự hiện diện của một alen alpha globin hoạt động hỗ trợ tổng hợp Hb F đủ để thai nhi sống sót trong thời kỳ mang thai và tổng hợp đủ Hb A để duy trì sự sống sau khi sinh, mặc dù có tỷ lệ mắc bệnh đáng kể.
Bệnh Hb H được xác định đồng thời bởi hai nhóm điều tra viên vào năm 1955. Những bệnh nhân bị ảnh hưởng bị thiếu máu hồng cầu nhỏ giảm sắc tố ở mức độ nghiêm trọng khác nhau; tế bào đích trên phết tế bào ngoại vi; tăng hồng cầu lưới; thể vùi trong một vài tế bào hồng cầu (RBC) được gọi là thể Heinz, nhiều tế bào khác có thể được tạo ra bằng cách ủ hồng cầu với thuốc nhuộm oxy hóa nhẹ như xanh cresyl rực rỡ, khả năng chống lại sự ly giải của dung dịch nhược trương và các dấu hiệu tan máu, bao gồm cả sự rút ngắn.
Một phát hiện quan trọng là sự hiện diện của một huyết sắc tố bất thường, chiếm tới 40% tổng lượng huyết sắc tố, có khả năng di động điện di nhanh hơn ở pH 8,6 so với Hb A. Khi đứng yên, Hb H chuyển sang màu nâu và kết tủa, tạo thành các khối trông hơi giống các thể vùi hồng cầu, một hiện tượng được nhấn mạnh khi bổ sung các chất oxy hóa nhẹ.
Trong bệnh mất đoạn Hb H, bệnh nhân chỉ được thừa hưởng một gen alpha globin duy nhất (a-/–). Ở dạng không xóa, bệnh nhân được thừa hưởng hai gen alpha globin từ cha hoặc mẹ, nhưng một trong số chúng mang khiếm khuyết không xóa, chẳng hạn như đột biến điểm (aa*/–, trong đó a* đại diện cho một biến thể chẳng hạn như Hemoglobin Constant Spring). Các đột biến alpha + phổ biến nhất tạo ra các huyết sắc tố không ổn định cao, làm tăng thêm gánh nặng cơ thể bao gồm hồng cầu.
* Alpha thalassemia thể nhẹ/tính trạng – Sự kế thừa hai gen alpha bình thường và hai gen alpha đột biến, được gọi là tính trạng alpha thalassemia thể nhẹ (alpha thalassemia minor) hoặc alpha thalassemia-1, có thể xảy ra với tình trạng dị hợp tử đối với kiểu gen alpha 0 (aa/–) hoặc dị hợp tử đối với gen alpha + kiểu gen (a-/a-). Những người này bị thiếu máu tối thiểu hoặc không có thiếu máu và hồng cầu nhỏ (thể tích tiểu cầu trung bình thấp (Mean Corpuscular Volume – MCV).
* Alpha thalassemia tối thiểu/người mang mầm bệnh thầm lặng – Di truyền ba gen alpha bình thường và một gen đột biến (aa/a-) được gọi là tính trạng alpha thalassemia tối thiểu (Alpha thalassemia minima), người mang gen thầm lặng, hoặc đặc điểm alpha thalassemia-2. Những cá nhân này thường không có bất thường về lâm sàng hoặc huyết học. Chẩn đoán chỉ có thể được thực hiện một cách đáng tin cậy thông qua phân tích ADN.
* Các biến thể alpha thalassemia phức tạp – Ngoài các biến thể được mô tả ở trên, các cá nhân có thể có các biến thể alpha 0 từ một bên cha mẹ và các biến thể alpha + từ bên cha mẹ kia.
Sự kết hợp của các biến thể huyết sắc tố
Sự phức tạp hơn nữa xảy ra khi một cá nhân mang sự kết hợp của các biến thể gen alpha và beta globin, hoặc sự kết hợp của bệnh thalassemia alpha hoặc beta với một biến thể huyết sắc tố khác như huyết sắc tố hình liềm hoặc Hb C.
Theo nguyên tắc chung, bệnh alpha thalassemia cộng với một biến thể beta globin có xu hướng cải thiện một phần mức độ nghiêm trọng của biến thể beta globin, trong khi bệnh beta thalassemia cộng với một biến thể beta globin như Hb S có xu hướng tạo ra kiểu hình lâm sàng của biến thể kia, thường là nhẹ. nhẹ hơn (thalassemia beta hình liềm) vì alen thalassemia làm giảm nồng độ huyết sắc tố nội bào, làm giảm bệnh hồng cầu hình liềm.
Một số biến thể cấu trúc cũng liên quan đến giảm sinh tổng hợp globin (ví dụ: Hb E, được liệt kê bên dưới) hoặc tích lũy do mất ổn định (ví dụ: Hb Terra Haute), trong trường hợp đó, sự đồng thời của alen beta thalassemia làm trầm trọng thêm kiểu hình lâm sàng.
* Alpha và beta thalassemia – Những người có sự di truyền đồng hợp tử của beta + thalassemia đồng hợp tử (beta + /beta +) và alpha thalassemia (-a/-a, –/aa, hoặc -a/aa) sẽ biểu hiện với mức độ lâm sàng trung bình. Việc giảm đồng thời cả chuỗi alpha và chuỗi beta làm giảm mức độ mất cân bằng so với những gì sẽ xảy ra nếu chỉ một hoặc một loại chuỗi khác bị ảnh hưởng.
* Sự tồn tại di truyền của huyết sắc tố thai nhi (fetal hemoglobin) – Một số biến thể beta globin dẫn đến việc sản xuất Hb F ở mức độ cao hơn, có thể đóng vai trò là đối tác giống như beta globin với alpha globin, dẫn đến sự mất cân bằng ít nghiêm trọng hơn về tỷ lệ globin alpha-beta trong thai nhi. những người mắc bệnh beta thalassemia.
* Bệnh thiếu máu beta thalassemia hình liềm (Sickle-beta thalassemia) – Sự di truyền đồng thời của đột biến hình liềm trên một alen beta globin và một đột biến bệnh thalassemia ở một alen beta globin khác dẫn đến bệnh hồng cầu hình liềm, mức độ nghiêm trọng của bệnh phụ thuộc vào bản chất của đột biến beta thalassemia (tức là liệu nó dẫn đến giảm [beta + ] hoặc không sản xuất [beta 0 ] beta globin).
* Hb E – Hb E là kết quả của đột biến beta globin làm giảm sản xuất beta globin. Dị hợp tử Hb E và một biến thể beta thalassemia chịu trách nhiệm cho một tỷ lệ lớn bệnh beta thalassemia nặng trên toàn thế giới [ 16 ]. Nó xảy ra với tần suất từ 3 đến 9 phần trăm ở Thái Lan. Kiểu hình lâm sàng không đồng nhất, với nồng độ hemoglobin dao động từ khoảng 3 đến 14 g/dL; sự đồng thừa kế của một biến thể alpha thalassemia có thể điều chỉnh kiểu hình. Hb E thường thấy ở Ấn Độ và Đông Nam Á. Kiểu hình dường như đặc biệt nghiêm trọng ở Sri Lanka
* Thalassemia trội (Dominant thalassemia) – Thalassemia trội đề cập đến một số biến thể beta thalassemia hiếm gặp đơn lẻ tạo ra hình ảnh bệnh ở trạng thái dị hợp tử, điển hình được đặc trưng bởi kiểu hình NTD beta thalassemia thể trung gian và đôi khi có hình ảnh lâm sàng phụ thuộc vào truyền máu [ 20 ] . Các biến thể trội trên nhiễm sắc thể thường này thường là các đột biến sai nghĩa ở exon 3 tạo ra các globin không ổn định và siêu không ổn định. Các biến thể beta thalassemia chiếm ưu thế này thường được mô tả là đột biến de novo trong các gia đình có nguồn gốc dân tộc phân tán. Phần lớn các trường hợp không được phát hiện bằng xét nghiệm thông thường, và do đó chúng cần tăng cường nhận thức lâm sàng và xét nghiệm chuyên biệt.
Không thể dự đoán hoàn toàn kiểu hình lâm sàng từ kiểu gen globin, vì các biến thể di truyền khác và các yếu tố môi trường có thể ảnh hưởng đến mức độ nghiêm trọng của bệnh.
* Bệnh alpha thalassemia mắc phải trong MDS — Những người mắc chứng rối loạn tạo máu vô tính như hội chứng loạn sản tủy (Myelodysplastic Syndrome – MDS) có thể phát triển bệnh alpha thalassemia mắc phải trong quần thể vô tính; đây còn được gọi là bệnh Hb H mắc phải.
Tài liệu tham khảo
- https://www.sciencedirect.com/topics/medicine-and-dentistry/sickle-cell-beta-thalassemia
- https://rarediseases.org/gard-rare-disease/sickle-beta-thalassemia/
- https://www.medicalnewstoday.com/articles/sickle-cell-beta-thalassemia
- https://thalassemia.com/genetics-inheritance.aspx#gsc.tab=0
- https://www.cancer.org/cancer/types/myelodysplastic-syndrome/about/what-is-mds.html