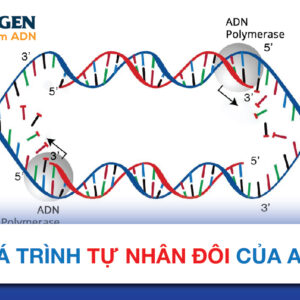
Gen HBB nằm trên nhiễm sắc thể 11, mang thông tin hướng dẫn để tạo ra beta-globin, một thành phần (tiểu đơn vị) của hemoglobin trong các tế bào hồng cầu. Vậy chi tiết chức năng của gen HBB là gì? Có những đột biến gen HBB gây bệnh nào? Tham khảo bài viết NOVAGEN chia sẻ sau để tìm hiểu chi tiết!

Nội dung:
1. Chức năng của gen HBB
Gen HBB cung cấp hướng dẫn để tạo ra một loại protein gọi là beta-globin. Beta-globin là một thành phần (tiểu đơn vị) của một loại protein lớn hơn gọi là hemoglobin, nằm bên trong các tế bào hồng cầu. Ở người lớn, hemoglobin thường bao gồm bốn tiểu đơn vị protein: hai tiểu đơn vị beta-globin và hai tiểu đơn vị của một loại protein gọi là alpha-globin, được sản xuất từ gen HBA . Mỗi tiểu đơn vị protein này được gắn (liên kết) với một phân tử chứa sắt gọi là heme; sắt ở trung tâm của mỗi heme có thể liên kết với một phân tử oxy. Hemoglobin trong các tế bào hồng cầu liên kết với các phân tử oxy trong phổi. Sau đó, các tế bào hồng cầu di chuyển qua mạch máu và cung cấp oxy cho các mô trên khắp cơ thể.
2. Đột biến gen HBB gây bệnh
2.1. Bệnh beta thalassemia
Có tới hàng trăm biến thể gen HBB (còn gọi là đột biến) gây ra bệnh beta thalassemia đã được tìm thấy. Hầu hết các biến thể này đều có liên quan đến sự thay đổi nucleotide đơn trong cấu trúc ADN tại vị trí gen HBB hoặc gần gen HBB. Các biến thể khác sẽ chèn hoặc xóa một số lượng nhỏ nucleotide trong gen HBB .
Các biến thể gen HBB làm giảm khả năng sản xuất beta-globin và sẽ gây ra tình trạng gọi là thalassemia beta-plus (β+). Các biến thể ngăn cản, không cho tế bào sản xuất bất kỳ beta-globin nào và sẽ gây ra bệnh thalassemia beta-zero (β0).
Nồng độ beta-globin thấp có thể ảnh hưởng đến quá trình sản xuất hemoglobin. Thiếu hemoglobin làm gián đoạn quá trình phát triển bình thường của các tế bào hồng cầu. Thiếu các tế bào hồng cầu trưởng thành có thể làm giảm đáng kể lượng oxy được cung cấp cho các mô. Thiếu oxy trong các mô của cơ thể có thể dẫn đến tình trạng kém phát triển, tổn thương cơ quan và các vấn đề sức khỏe khác liên quan đến bệnh beta thalassemia.
2.2. Bệnh Methemoglobin huyết, loại beta-globin
Các biến thể trong gen HBB cũng là nguyên nhân gây ra bệnh methemoglobinemia, loại beta-globin, một tình trạng làm thay đổi hemoglobin trong các tế bào hồng cầu. Các biến thể này thường ảnh hưởng đến vùng protein liên kết với heme. Để hemoglobin liên kết với oxy, sắt trong phân tử heme cần phải ở dạng gọi là sắt II (Fe2+). Sắt trong heme có thể chuyển thành dạng sắt khác gọi là sắt III (Fe3+), không thể liên kết với oxy. Hemoglobin chứa sắt III được gọi là methemoglobin và không thể vận chuyển oxy đến các mô của cơ thể một cách hiệu quả.
Ở những người mắc bệnh methemoglobinemia, loại beta-globin, các biến thể trong gen HBB làm thay đổi protein beta-globin và khiến sắt heme chuyển từ sắt (II) sang sắt (III). Hemoglobin bị thay đổi này làm cho máu có màu nâu và khiến da, môi, móng tay có màu hơi xanh (tím tái). Các dấu hiệu và triệu chứng của bệnh methemoglobinemia, loại beta-globin thường chỉ giới hạn ở chứng tím tái, không gây ra bất kỳ vấn đề sức khỏe nào. Tuy nhiên, trong một số trường hợp hiếm gặp, bệnh methemoglobinemia, loại beta-globin nghiêm trọng có thể gây đau đầu, yếu và mệt mỏi.
2.3. Bệnh hồng cầu hình liềm
Thiếu máu hồng cầu hình liềm (còn gọi là bệnh hồng cầu hình liềm đồng hợp tử hoặc bệnh HbSS) là dạng bệnh hồng cầu hình liềm phổ biến nhất. Dạng bệnh này do một biến thể đặc biệt trong gen HBB gây ra, dẫn đến sản xuất ra phiên bản beta-globin bất thường gọi là hemoglobin S (HbS). Ở những người mắc tình trạng này, hemoglobin S thay thế cả hai tiểu đơn vị beta-globin trong hemoglobin.
Biến thể gây ra hemoglobin S thay đổi một khối xây dựng protein đơn lẻ (axit amin) trong beta-globin. Cụ thể, axit amin glutamic được thay thế bằng axit amin valine ở vị trí 6 trong beta-globin, được viết là Glu6Val (E6V). Việc thay thế axit glutamic bằng valine khiến các tiểu đơn vị hemoglobin S bất thường dính lại với nhau và tạo thành các phân tử dài, cứng làm cong các tế bào hồng cầu thành hình lưỡi liềm. Các tế bào hình lưỡi liềm chết quá sớm, có thể dẫn đến tình trạng thiếu hồng cầu (thiếu máu). Các tế bào hình lưỡi liềm cứng có thể chặn các mạch máu nhỏ, gây đau dữ dội và tổn thương các cơ quan.
Các biến thể trong gen HBB cũng có thể gây ra các bất thường khác trong beta-globin, dẫn đến các loại bệnh hồng cầu hình liềm khác. Trong các loại bệnh hồng cầu hình liềm khác này, chỉ một tiểu đơn vị beta-globin được thay thế bằng hemoglobin S. Tiểu đơn vị beta-globin khác được thay thế bằng một phiên bản beta-globin khác, chẳng hạn như hemoglobin C (HbC) hoặc hemoglobin E (HbE).
Trong bệnh hemoglobin SC (HbSC), các tiểu đơn vị beta-globin được thay thế bằng hemoglobin S và hemoglobin C. Hemoglobin C xuất hiện khi axit amin lysine thay thế axit glutamic ở vị trí 6 trong beta-globin (viết là Glu6Lys hoặc E6K). Mức độ nghiêm trọng của bệnh hemoglobin SC khác nhau, nhưng có thể nghiêm trọng như bệnh thiếu máu hồng cầu hình liềm. Hemoglobin E xuất hiện khi axit glutamic được thay thế bằng lysine ở vị trí 26 trong beta-globin (viết là Glu26Lys hoặc E26K). Trong một số trường hợp, hemoglobin E có mặt cùng với hemoglobin S. Trong những trường hợp này, một người có thể có các dấu hiệu và triệu chứng nghiêm trọng hơn hoặc tương tự như những dấu hiệu và triệu chứng thấy ở những người bị thiếu máu hồng cầu hình liềm, chẳng hạn như các cơn đau, thiếu máu và chức năng lách bất thường.
Một tình trạng khác, được gọi là hemoglobin S-beta thalassemias (HbSBetaThal), xảy ra khi các biến thể dẫn đến hemoglobin S và beta thalassemia (được mô tả ở trên) xảy ra cùng nhau. Các biến thể kết hợp bệnh hồng cầu hình liềm với beta-zero thalassemia (β0) dẫn đến bệnh nghiêm trọng, trong khi bệnh hồng cầu hình liềm kết hợp với beta-plus thalassemia (β+) thường nhẹ hơn.
2.4. Các rối loạn khác
Hàng trăm biến thể đã được xác định trong gen HBB. Những biến thể này dẫn đến việc sản xuất ra các phiên bản beta-globin khác nhau. Một số biến thể không gây ra dấu hiệu hoặc triệu chứng đáng chú ý và được tìm thấy khi xét nghiệm máu vì những lý do khác, trong khi các biến thể gen HBB khác có thể ảnh hưởng đến sức khỏe. Hai trong số các phiên bản thay thế phổ biến nhất của beta-globin là hemoglobin C và hemoglobin E (được mô tả ở trên).
Hemoglobin C, do sự thay đổi Glu6Lys trong beta-globin, phổ biến hơn ở những người gốc Tây Phi so với những nhóm dân số khác. Những người có hai tiểu đơn vị hemoglobin C trong hemoglobin của họ, thay vì beta-globin bình thường, mắc một tình trạng nhẹ gọi là bệnh hemoglobin C. Tình trạng này thường gây ra tình trạng thiếu máu mãn tính, trong đó các tế bào hồng cầu bị phá vỡ sớm.
Hemoglobin E, do sự thay đổi Glu26Lys trong beta-globin, thường gặp nhất ở nhóm dân số Đông Nam Á. Khi một người có hai tiểu đơn vị hemoglobin E trong hemoglobin của họ thay vì beta-globin, một bệnh thiếu máu nhẹ gọi là bệnh hemoglobin E có thể xảy ra. Trong một số trường hợp, các biến thể tạo ra hemoglobin E và beta thalassemia (được mô tả ở trên) được tìm thấy cùng nhau. Những người có sự kết hợp hemoglobin này có thể có các dấu hiệu và triệu chứng từ thiếu máu nhẹ đến thalassemia nặng.
Tham khảo: Medlineplus