Hội chứng Vi mất đoạn (Microdeletion syndrome) là tên gọi chung cho các hội chứng liên quan đến đột biến mất hoặc xóa một đoạn gen nhỏ tại những vùng đặc biệt của nhiễm sắc thể, gây ra một số khiếm khuyết cho cơ thể.
Nội dung:
1. Thế nào là vi mất đoạn? Có những hội chứng vi mất đoạn nào?
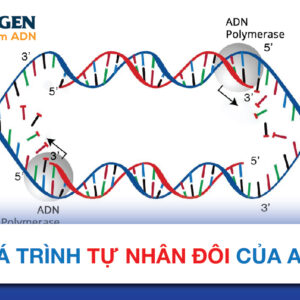
Vi mất đoạn (Microdeletion) là tình trạng mất hay xóa một đoạn gen siêu nhỏ (có kích thước chỉ 1-3 Mb) tại các vị trí đặc biệt của nhiễm sắc thể (thường là các gen tiếp giáp trên nhiễm sắc thể). Việc xét nghiệm các hội chứng vi mất đoạn được thực hiện bằng phương pháp lai huỳnh quang tại chỗ (FISH) và có thể sàng lọc trước sinh thông qua xét nghiệm NIPT với gói NIPT Premium chuyên sâu nhất
Đột biến vi mất đoạn tại các vùng quan trọng của gen sẽ thường dẫn đến những thay đổi kiểu hình đặc trưng do những biểu hiện của các phân đoạn gen cụ thể bị thiếu hoặc sai lệch, dẫn tới các hội chứng vi mất đoạn cụ thể.
Tên của các hội chứng vi mất đoạn được đặt theo đoạn gen bị mất trên nhiễm sắc thể với số đầu tiên là số của bộ nhiễm sắc thể, còn cụm chữ và số đằng sau là vị trí của đoạn gen bị mất. “p” và “q” là hai “cánh tay” của nhiễm sắc thể, trong đó nhánh dài là nhánh q, nhánh ngắn là nhánh p.
Ví dụ: Với Hội chứng mất đoạn 8q22.1 sẽ được hiểu là mất đoạn q22.1 trên nhiễm sắc thể số 8.
a. Những hội chứng vi mất đoạn phổ biến
- Mất đoạn 15q11-13 có nguồn gốc từ mẹ (Hội chứng Angelman)
- Mất đoạn 15q11-13 có nguồn gốc từ cha (Hội chứng Prader–Willi)
- Mất đoạn 7q11.23 (Hội chứng Williams)
- Mất đoạn 4p (Hội chứng Wolf–Hirschhorn)
- Mất đoạn trên NST số 5 (Hội chứng Cri du chat tiếng mèo kêu)
- Mất đoạn 22q11.2 (Hội chứng DiGeorge)
b. Những hội chứng vi mất đoạn khác đã được ghi nhận y khoa
- Hội chứng vi mất đoạn 1q36
- Hội chứng vi mất đoạn 1q21.1
- Hội chứng vi mất đoạn 2q15-16.1
- Hội chứng vi mất đoạn 2q23.1
- Hội chứng vi mất đoạn 2q37
- Hội chứng vi mất đoạn 3p
- Hội chứng vi mất đoạn 3q29
- Hội chứng vi mất đoạn 5q35 (Sotos syndrome)
- Hội chứng vi mất đoạn 6p25
- Hội chứng vi mất đoạn 8p23.1
- Hội chứng vi mất đoạn 8q22.1 (Nablus mask-like facial syndrome)
- Hội chứng vi mất đoạn 8q24.11 (Langer – Giedion syndrome hay trichorhinophalangeal syndrome type II)
- Hội chứng vi mất đoạn 9q22
- Hội chứng vi mất đoạn 9q34.3
- Hội chứng vi mất đoạn 11q13 (WAGR syndrome)
- Hội chứng vi mất đoạn 11q11.2 (Potocki – Shaffer syndrome)
- Hội chứng vi mất đoạn 11q24.1 (Jacobsen syndrome)
- Hội chứng vi mất đoạn 13q14 (retinoblastoma syndrome)
- Hội chứng vi mất đoạn 15q11.2 (BP1-2)
- Hội chứng vi mất đoạn 15q13.3
- Hội chứng vi mất đoạn 15q15.3
- Hội chứng vi mất đoạn 15q24
- Hội chứng vi mất đoạn 16p13.11
- Hội chứng vi mất đoạn 17p13.3
- Hội chứng vi mất đoạn 17p1.2
- Hội chứng vi mất đoạn 17q12
- Hội chứng vi mất đoạn 17q21.31
- Hội chứng vi mất đoạn 18p
- Hội chứng vi mất đoạn 20p11
2. Một số hội chứng vi mất đoạn phổ biến
Dưới đây là thông tin về một số hội chứng vi mất đoạn phổ biến, bao gồm tổng quan, dấu hiệu, triệu chứng và tỷ lệ mắc.
2.1. Hội chứng Angelman (mất đoạn 15q11-13 từ mẹ)
Hội chứng Angelman là một rối loạn di truyền phức tạp chủ yếu ảnh hưởng đến hệ thần kinh, xảy ra bởi mất đoạn 15q11-13 từ mẹ làm vô hiệu hóa gen UBE3A.
Các đặc điểm đặc trưng của tình trạng này bao gồm chậm phát triển, khuyết tật trí tuệ, khiếm khuyết về giọng nói nghiêm trọng và các vấn đề về vận động và thăng bằng (rối loạn vận động).
Hầu hết trẻ em bị ảnh hưởng cũng bị co giật tái phát (động kinh) và đầu nhỏ (tật đầu nhỏ). Sự chậm phát triển trở nên đáng chú ý khi trẻ được 6 đến 12 tháng tuổi và các dấu hiệu và triệu chứng phổ biến khác thường xuất hiện ở thời thơ ấu.
Tỷ lệ mắc: 1/12.000-20.000
2.2. Hội chứng Prader–Willi
Hội chứng Prader-Willi là một tình trạng di truyền phức tạp ảnh hưởng đến nhiều bộ phận của cơ thể, xảy ra bởi mất đoạn 15q11-13 từ cha.
Ở trẻ sơ sinh, tình trạng này được đặc trưng bởi trương lực cơ yếu (giảm trương lực), khó khăn khi ăn, chậm phát triển và chậm phát triển. Bắt đầu từ thời thơ ấu, những người bị ảnh hưởng sẽ phát triển cơn đói cực độ, dẫn đến ăn quá nhiều mãn tính (ăn nhiều) và béo phì. Một số người mắc hội chứng Prader-Willi, đặc biệt là những người béo phì, cũng mắc bệnh tiểu đường loại 2 (dạng tiểu đường phổ biến nhất).
Những người mắc hội chứng Prader-Willi thường bị suy giảm trí tuệ từ nhẹ đến trung bình và khuyết tật học tập. Các vấn đề về hành vi cũng phổ biến, bao gồm các cơn nóng giận, bướng bỉnh và hành vi cưỡng chế như cào da. Rối loạn giấc ngủ cũng có thể xảy ra.
Các đặc điểm bổ sung của tình trạng này bao gồm các đặc điểm khuôn mặt đặc biệt như trán hẹp, mắt hình quả hạnh và miệng hình tam giác; vóc dáng thấp; và bàn tay và bàn chân nhỏ.
Một số người mắc hội chứng Prader-Willi có làn da trắng bất thường và tóc sáng màu. Cả nam giới và nữ giới bị ảnh hưởng đều có bộ phận sinh dục kém phát triển. Tuổi dậy thì bị chậm lại hoặc không đầy đủ, và hầu hết những người bị ảnh hưởng đều không có khả năng sinh con (vô sinh).
Tỷ lệ mắc: 1/10.000-30.000
2.3. Hội chứng Williams
Hội chứng Williams là một rối loạn phát triển ảnh hưởng đến nhiều bộ phận của cơ thể.
Tình trạng này được đặc trưng bởi khuyết tật trí tuệ hoặc vấn đề học tập từ nhẹ đến trung bình, đặc điểm tính cách độc đáo, các đặc điểm khuôn mặt đặc biệt và các vấn đề về tim và mạch máu (tim mạch).
Những người mắc hội chứng Williams thường gặp khó khăn với các nhiệm vụ thị giác-không gian như vẽ và lắp ráp câu đố, nhưng họ có xu hướng làm tốt các nhiệm vụ liên quan đến ngôn ngữ nói, âm nhạc và học bằng cách lặp lại (ghi nhớ thuộc lòng). Những người bị ảnh hưởng có tính cách hướng ngoại, hòa đồng và có xu hướng cực kỳ quan tâm đến người khác. Rối loạn thiếu chú ý (ADD), các vấn đề về lo lắng và ám ảnh sợ hãi là phổ biến ở những người mắc chứng rối loạn này.
Trẻ nhỏ mắc hội chứng Williams có các đặc điểm khuôn mặt đặc biệt bao gồm trán rộng, bọng mắt, sống mũi phẳng, má đầy đặn và cằm nhỏ. Nhiều người bị ảnh hưởng có các vấn đề về răng miệng như răng nhỏ, thưa thớt, mọc lệch hoặc mất. Trẻ lớn hơn và người lớn thường có khuôn mặt dài hơn với miệng rộng và môi đầy đặn.
Các dấu hiệu và triệu chứng bổ sung của hội chứng Williams bao gồm các bất thường của mô liên kết (mô hỗ trợ các khớp và cơ quan của cơ thể) như các vấn đề về khớp và da mềm, lỏng lẻo.
Những người bị ảnh hưởng cũng có thể có nồng độ canxi trong máu tăng (tăng canxi huyết) ở trẻ sơ sinh, chậm phát triển, các vấn đề về phối hợp và vóc dáng thấp.
Các vấn đề y tế liên quan đến thị lực hoặc thính lực, bao gồm nhạy cảm với âm thanh (hyperacusis), thường liên quan đến hội chứng Williams. Ngoài ra, các vấn đề về đường tiêu hóa và hệ tiết niệu cũng có thể xảy ra. Béo phì hoặc tiểu đường có thể phát triển ở tuổi trưởng thành.
Tỷ lệ mắc: 1/7.500-18.000
2.4. Hội chứng Wolf–Hirschhorn
Hội chứng Wolf-Hirschhorn là hội chứng gây nên do mất một đoạn gen nhỏ cuối nhánh ngắn (p) của nhiễm sắc thể số 4, dẫn tới mất các gen NSD2, LETM1 và MSX1 dẫn đến khuyết tật trí tuệ và bất thường về thể chất nghiêm trọng.
Các đặc điểm chính của rối loạn này bao gồm các đặc điểm khuôn mặt đặc trưng, chậm phát triển và tăng trưởng, khuyết tật trí tuệ và co giật.
Hầu như tất cả bệnh nhân mắc chứng rối loạn này đều có các đặc điểm khuôn mặt đặc biệt, bao gồm sống mũi rộng, mắt to và lồi, và trán cao. Ngoài ra, những người bị ảnh hưởng có thể có các đặc điểm khuôn mặt không cân xứng và đầu nhỏ bất thường (tật đầu nhỏ).
Những người mắc hội chứng Wolf-Hirschhorn bị chậm phát triển và tăng trưởng. Quá trình tăng trưởng chậm bắt đầu trước khi sinh và trẻ sơ sinh bị ảnh hưởng có xu hướng gặp vấn đề về ăn uống và tăng cân (không phát triển). Trẻ cũng bị yếu cơ (hạ trương lực cơ) và cơ kém phát triển. Các kỹ năng vận động như ngồi, đứng và đi bộ bị chậm đáng kể. Hầu hết trẻ em và người lớn mắc chứng rối loạn này cũng có vóc dáng thấp bé.
Khuyết tật trí tuệ có thể từ nhẹ đến nặng ở những người mắc hội chứng Wolf-Hirschhorn. So với những người mắc các dạng khuyết tật trí tuệ khác, kỹ năng xã hội của họ rất mạnh, nhưng kỹ năng giao tiếp bằng lời nói và ngôn ngữ có xu hướng yếu hơn. Hầu hết trẻ em bị ảnh hưởng cũng bị co giật, có thể kháng trị. Các cơn co giật có xu hướng biến mất theo tuổi tác.
Tỷ lệ mắc: 1/50.000
2.5. Hội chứng Cri du chat (tiếng mèo kêu)
Hội chứng Cri-du-chat (tiếng mèo kêu), còn được gọi là hội chứng 5p- , là một tình trạng nhiễm sắc thể xảy ra khi một đoạn nhiễm sắc thể số 5 bị thiếu, dẫn tới mất nhiều gen trên nhánh ngắn của nhiễm sắc thể số 5. Các nhà nghiên cứu tin rằng việc mất một gen cụ thể, CTNND2 có liên quan đến khuyết tật trí tuệ nghiêm trọng ở một số người mắc tình trạng này
Trẻ sơ sinh mắc tình trạng này thường có tiếng khóc the thé giống như tiếng mèo kêu. Rối loạn này được đặc trưng bởi khuyết tật trí tuệ và chậm phát triển, kích thước đầu nhỏ (tím cụt), cân nặng khi sinh thấp và trương lực cơ yếu (hạ huyết áp) ở trẻ sơ sinh.
Những cá nhân bị ảnh hưởng cũng có các đặc điểm khuôn mặt đặc biệt, bao gồm mắt cách xa nhau (hypertelorism), tai thấp, hàm nhỏ và khuôn mặt tròn. Một số trẻ mắc hội chứng cri-du-chat được sinh ra với khuyết tật tim.
2.6. Hội chứng DiGeorge
Hội chứng DiGeorge (hay hội chứng mất đoạn 22q11.2) xảy ra khi mất một đoạn nhỏ của nhiễm sắc thể 22 tại vị trí q11.2.
Các đặc điểm của hội chứng này rất khác nhau, ngay cả giữa những thành viên bị ảnh hưởng trong cùng một gia đình.
Những người mắc hội chứng mất đoạn 22q11.2 thường có bất thường về tim thường xuất hiện từ khi sinh ra, nhiễm trùng tái phát do các vấn đề về hệ thống miễn dịch và các đặc điểm khuôn mặt đặc biệt.
Ở những người bị ảnh hưởng, các cơ tạo thành vòm miệng (vòm miệng) có thể không đóng hoàn toàn, mặc dù mô bao phủ chúng đã đóng, dẫn đến tình trạng gọi là hở hàm ếch dưới niêm mạc. Vòm miệng bất thường thường cong nhiều và có thể có một vết nứt ở vạt mô mềm treo ở phía sau miệng (lưỡi gà chẻ đôi). Hở hàm dưới niêm mạc cũng có thể cản trở lời nói bình thường bằng cách khiến không khí thoát ra khỏi mũi trong khi nói, dẫn đến giọng nói phát ra âm thanh mũi.
Những người bị ảnh hưởng cũng có thể gặp các vấn đề về hô hấp, bất thường về thận, nồng độ canxi trong máu thấp (có thể dẫn đến co giật), giảm tiểu cầu trong máu (giảm tiểu cầu), khó khăn đáng kể khi ăn, các vấn đề về đường tiêu hóa và mất thính lực. Có thể có những khác biệt về xương, bao gồm vóc dáng thấp nhẹ và ít gặp hơn là bất thường ở xương sống.
Nhiều trẻ mắc hội chứng mất đoạn 22q11.2 bị chậm phát triển, bao gồm chậm phát triển và chậm nói, và một số trẻ bị khuyết tật trí tuệ nhẹ hoặc khuyết tật học tập. Những người lớn tuổi bị ảnh hưởng gặp khó khăn trong việc đọc, thực hiện các nhiệm vụ liên quan đến toán học và giải quyết vấn đề. Ngoài ra, trẻ em bị ảnh hưởng có nhiều khả năng mắc chứng rối loạn thiếu tập trung/tăng động (ADHD) và các tình trạng phát triển như rối loạn phổ tự kỷ ảnh hưởng đến giao tiếp và tương tác xã hội.
Tỷ lệ mắc: ¼.000
Nguồn: NOVAGEN, Medline